Introduction
Bactrocera tryoni is a species of fruit fly and a horticultural pest commonly found in Queensland and Victoria. Like Drosophila melanogaster, B. tyroni is a model organism that the experiment employed to demonstrate gene mapping of the white gene using visible and molecular markers. B. tryoni specimens offer an appropriate model organism for gene mapping because they exhibit sympatric speciation, submit to forced copulation, and exist as pure strains (Gilchrist et al. 2014). A molecular technique, gene mapping entails the identification of genetic markers, the location of genes, genetic linkages and distances between genes.
Gene mapping of B. tyroni is significant in determining the locus of the white gene and the pattern of inheriting phenotypes and genotypes. Single nucleotide polymorphisms (SNPs) and simple sequence repeats (SSRs) are two forms of molecular markers applied in mapping the white gene in B. tyroni. The white gene is a 680bp polymorphic exon with allele a having two restriction sites and allele b having one restriction site for Rsal (GTWC) (Choo et al. 2017). Therefore, the laboratory report aims to amplify the white gene, conduct restriction enzyme digest, and analyze restriction fragments with the overall objective of creating a gene map of visible and molecular markers.
Materials and Methods
We used a parental cross to obtain F1 progeny and a male backcross to get G2 progeny. The parental cross occurred between one stock of flies with homozygous wild-type white marks and homozygous recognition sites (wm+/wm+ Ra /Ra) and another stock with homozygous mutant white marks and homozygous recognition sites (wm/wm Rb/Rb). We performed male backcross to obtain dominant white marks and a higher proportion of Rsal recognition sites in the G2 progeny. We isolated DNA samples from parental, F1, and G2 flies and labeled them according to their numbers and phenotypes. Each pair of students participated in the amplification of white genes using a thermal cycler. The final concentration of PCR master mix components were 0.5 mM dNTPs, 3 mM MgCl2, 67 mM Tris-HCL (pH 8), 16.6 mM [NH]SO4, 0.2 mg/ml gelatin and 0.45% Triton X-100, while primer mix and thermostable DNA polymerase had 0.35 units/μl and 12.5 μM, respectively. We prepared a total reaction volume of PCR (25 μl), which consisted of 18 μl of PCR master mix, 2 μl of primer mix, 3 μl of DNA, template and 2 μl of Taq polymerase.
We placed reactants in a thermal cycler and set the PCR conditions as initial denaturation at 94°C for 3 minutes, denaturation at 94°C for 1 minute, annealing at 60°C for 1 minute, extension at 72°C for 1 minute, and a final extension at 72°C for 5 minutes. The PCR ran for three hours, and its products were obtained and stored under a freezing condition (-20°C). For each PCR product, we digested half the volume with Rsal while the remaining half of the volume remained undigested. In the restriction digest, sterile Mili-Q water, 10X restriction buffer, and PCR master mix consisting of the final concentration of 500 nM NaCl, 100 mM DTT, 100 mM MgCl2, and 100 mM Tris-HCl were reagents. We set up the restriction enzyme digest in a total volume of 20 μl consisting of 6 μl mili-Q water, 10X buffer, 10 μl, PCR product, and 2 μl of Rsal 1.75/μl. Next, we mixed the digestion contents gently and incubated them for 1 hour in a water bath at 37°C.
The experiment used gel tanks, a power pack, a UV transilluminator, and a video camera as the apparatus in agarose gel electrophoresis to determine the sizes of Rsal fragments. We loaded 20 μl of digested PCR products and 12 μl of undigested PCR products together with their controls (P1, P2, and F1) in their respective wells. Gel electrophoresis ran at 180 volts until the dye front reached close to the next wells. We transferred the gel to the imaging system and took a photograph to show the results of the banding pattern.
Results
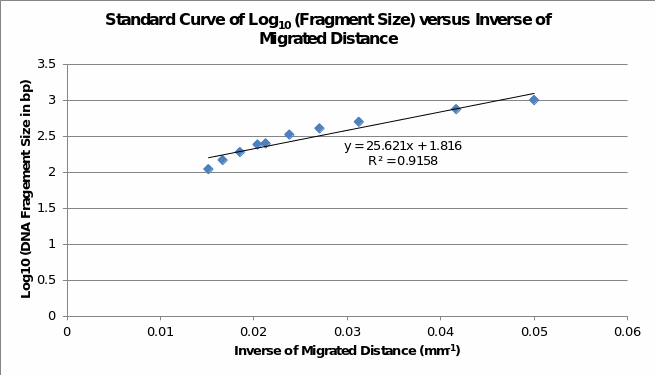
Table 1 and Table 2 show the estimated fragment sizes of digested and undigested PCR products, respectively. Although fragments on the gel image approximate the expected sizes, the use of a standard curve exhibits substantial variation because fragment lengths of undigested DNA are less than 680bp.
Table 1: Estimated Fragment Sizes of Undigested PCR Product.
Table 2: Estimated Fragment Sizes of Digested PCR Product.
Analysis of phenotypes and microsatellites (Table 3) shows that visible markers and the white gene markers link to wm loci, Bt1, Bt2, and Bt7. In the Bt2 loci, wild-type alleles link long alleles and contribute to the occurrence of homozygous Ra RFLP, whereas white markers link to short alleles and exhibit homozygous Rb RFLP. Moreover, Bt2 and Bt7 link to white RFLP because their short alleles express homogenous Rb phenotypes, while heterogeneous (SL) and homogenous alleles (LL) express Ra RFLP. Thus, the white and wm genes map to chromosomes two and five.
Table 3: Phenotypes of White Markers and Microsatellites in Various Chromosomes and Gene Loci.
Discussion
Analyzing the results obtained from the undigested and digested PCR products reveals important information that maps white and wm genes maps onto the Bt1, Bt2, and Bt7 loci of microsatellites. These findings are consistent with those of Choo et al. (2017), which demonstrated that the white gene determines the expression of the phenotype of white eyes in B. tyroni. Since B. tryoni has a series of microsatellites, it is difficult to establish the sequence of genes on the linked chromosomes. Genome-wide sequencing or target sequencing would enable accurate and precise mapping of the white and wm genes in B. tyroni (Costain, Kannu & Bowdin 2018; Gilchrist et al. 2014). Analysis of the sequences provides accurate information about the location of molecular markets, trait loci, and variations.
Examining the methodology shows that gel electrophoresis did not render clear and accurate banding patterns, resulting in an underestimation of fragment sizes of DNA. Reduction of the voltage and time for running gel would ensure the formation of sharp bands and prevent overrun of short sequences. The bulk segregant analysis is a modern method that could complement the techniques employed in this research; Pool (2016) has employed it in mapping pigmentation genes in D. melanogaster. Therefore, future research should focus on elucidating the mechanism and pattern of inheriting the white gene and related molecular markers.
The report has described the use of simple molecular techniques, such as PCR, restriction digest, and gel electrophoresis in mapping the white gene in B. tryoni. The findings show that visible white markers and white genes closely link to the Bt2, Bt2, and Bt loci of microsatellites.
Supplementary Information


Reference List
Choo, A, Crisp, P, Saint, R, O’Keefe, LV & Baxter, SW 2017, ‘CRISPR/Cas9‐mediated mutagenesis of the white gene in the tephritid pest Bactrocera tryoni’, Journal of Applied Entomology, vol. 142, no. 1-2, pp. 52-58.
Costain, G, Kannu, P & Bowdin, S 2018, ‘Genome-wide sequencing expands the phenotypic spectrum of EP300 variants’, European Journal of Medical Genetics, vol. 63, no. 3, pp. 125-129.
Gilchrist, AS, Shearman, DC, Frommer, M, Raphael, KA, Deshpande, NP, Wilkins, MR et al. 2014, ‘The draft genome of the pest tephritid fruit fly Bactrocera tryoni: resources for the genomic analysis of hybridising species’, BMC Genomics, vol. 15, no. 1, pp. 15(1), 1-16.
Pool, JE 2016, ‘Genetic mapping by bulk segregant analysis in Drosophila: experimental design and simulation-based inference’, Genetics, vol. 204, no. 3, pp. 1295-1306.