Introduction
This document is a laboratory report on a genetic engineering experiment to transform the Escherichia coli bacteria with the DNA that encodes for the Green Fluorescent Protein (GFP) isolated from Aequorea victoria. A. victoria, otherwise known as the jellyfish, is native to the eastern Pacific Ocean. The GFP gene is isolated from the jellyfish. The objective of this experiment is to understand the process and importance of the genetic transformation of bacteria in real time with the aid of extrachromosomal DNA, alternatively referred to as plasmids. The central hypothesis is that once the pGLO plasmid encoding for GFP transforms in the bacterial cells, the trait of fluorescence will be induced.
The genetic phenomenon of transformation has been instrumental in expanding the scientific understanding the molecular foundation of genes as well as providing scientists with the ability to alter phenotypes. First pioneered by Frederick Griffith, the artificial insertion of exogenous DNA into prokaryotic and eukaryotic cells has evolved from the imprecise science of yesteryears to a technique routinely brought to bear in modern-day activities.
Artificial transformation is usually achieved by use of vectors referred to as pplasmids, which are small circular DNA structures capable of autonomous replication. Like viruses, plasmids are not considered to be a form of life as they require a living host in order to replicate.
Genetic transformation has applications in scientific study and industry (Gardner & Godrick, 2011). Plasmids are used in medical research to create rat disease model, in human gene therapy and the mass-production of human hormones such as insulin. Other uses include the design of genetically modified organisms and use as a visual marker. Another unique example of a real-world application is combating malaria by creating mosquitoes with green fluorescent gonads. It follows that understanding the biological, biochemical and genetic processes occurring during transformation is a valuable skill for geneticists.
Method
Prior to handling the live bacterial cultures, sterile procedures were used to protect the integrity of the culture and safeguard individual safety. UV goggles and disposal bags were also used. The methodology is derived from the laboratory manual.
The closed test tubes were labeled as +pGLO and -pGLO and placed in a tube rack (Snustad &Simmons, 2011). 250 µL of CaCl2 was then added to each opened tube using sterile pipettes. The significance of adding CaCl2 solution was so as to pre-incubate the E. coli by weakening the cell membranes. The Ca++ neutralizes DNA phosphate and membrane phospholipid anions (Matthews, van Holde & Ahem, 2000). The tubes were then placed on crushed ice. Crsuhed ice is preferred as it maximizes the transformation efficiency. Icing decelerates cell membrane degradation. Using sterile loops, single colonies of the bacteria from the starter kit were twirled into both tubes and stirred by spinning them between the index finger and thumb until the colonies entirely dispersed. An examination of the solution containing the pGLO DNA was done under UV light.
A sterile loop inserted into the plasmid DNA tube had a film across the ring. Appearance of this film is critical as it indicated successful plasmid DNA transfer. 10 µL of pGLO was then pipetted into the +pGLO tube. The tubes were then promptly returned to the ice to incubate for 10 minutes, with care taken to ensure their bottom ends were in contact with the ice. Prompt icing provides cellular recovery for optimal β-lactamase expression before streaking on the ampicillin culture. This recovery period can be amplified by a magnitude of 10 after an overnight incubation ((Kim & et al, 2010).
As the tubes incubated on the ice, four (4) agar plates containing Luria-Bertani (LB) broth were labeled on the bottom as opposed to on their lids. The four plates were: +pGLO LB/AMP, +pGLO LB/AMP/ARA, -pGLO LB/AMP, and -pGLO LB. The arabinose contained in the agar trigger DNA transcription in order to activate arabinose-digesting enzymes (arabinose operon). Since the plasmids have displaced some of the genes resposnsible for arabinose activity, these genes are inadvertently activated as well, leading to the glowing.
The tubes still on the rack holder were then heat treated by transferring them into a hot water bath at 42ºC for 50 seconds and then rapidly returned to ice for a further 2 minutes. The heat shock is significant as it will degrade the cellular membranes and increase their permeability (Matthews, van Holde & Ahem, 2000). A favorable distortion in permeability gives the plasmids access to the intercellular matrix.
The tubes were then placed on a bench at room temperature, opened, 250 microliters of the LB nutrient added using the sterile pipettes and incubated for 10 minutes. The bacteria were then evenly plated onto the different agar plates using sterile loops and placed in a 37ºC incubator overnight. The nutrient broth in these agar plates enables β-lactamase expression. Β-lactamase inactivates the ampicillin in the +pGLO LB/AMP/ARA plates. Observations were then documented under both ambient and UV light (Snustad &Simmons, 2011).

Results and Analysis
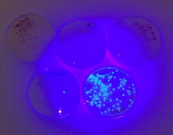
Bacterial lawns were observed the LB plate with -pGLO and the LB/AMP plate with +pGLO. Bacterial colonies grew on these plates. No bacterial colonies were found on the LB/AMP plate with –pGLO. Bio-luminant colonies were seen on the LB/AMP/ARA plate with +pGLO.
Fig. 2: Results from the bacterial cultures
The total amount of DNA used can be calculated as follows:
DNA (µg) = DNA concentration (µg/µl) X
(Volume of DNA in µl),
Hence DNA= 10 x 0.08= 0.8µl.
In addition,
Fraction of DNA used = Volume spread on
LB/AMP/AR plate
Total volume in stock
Tube,
To give a total of 0.2.
Further analysis requires to solve for pGLO spread,
pGLO spread (µg) = Total amount of DNA
used X Fraction of DNA,
To give 0.16.
These figures are essential for calculating,
Transformation efficiency = Total Number of
Transformant Colonies on the Culture Plates
Amount of DNA streaked on
the plates,
To give 280/0.16= 1,750/µg or 1.75 X 103 transformants/µg.
The calculated efficiency lies within the standard limits of efficiency.
Discussion
The hypothesis of this paper has been proven: upon addition of the pGLO plasmid, E. coli bacteria genetically transformed. As the individual results from the plates showed, the plate without the plasmid did not exhibit bioluminescence (glowing) while the plate with the plasmid developed ampicillin resistance and fluorescence (glowing).
Lapses in the experimental design and protocol, however, led to incorrect results. These errors can be occasioned by slow ice bath transitions, generous timing for the hot water baths, non-sterile procedures and the insufficient mixture of DNA with plasmids..
The utility of gene transformation is being explored in malaria containment, biosynthesis of medicine and replacement hormones, agriculture and chemical engineering, among others.
Conclusions
In conclusion, the experiment proved the hypothesis was right though an unknown experimental lapse was made. The E. coli was antibiotic resistant, and its luminescence was sustained by the arabinose. Artificial transformation was explored and its technique proven to work.
References
Gardner K., and Godrick E. (2011). Principles of Biology II. Hayden-McNeil Publishing, Plymouth, MI. Module 6, pp. 119-128.
Kim, J. Y.; Wang, Y.; Park, M. S.; Ji, G. E. (2010). Improvement of Transformation Efficiency through In-Vitro Methylation and SacII Site
Mutation of Plasmid Vector in Bifidobacterium longum MG1. Journal of Microbiology and Biotechnology 20(6):1022-1026.
Mathews, C. K.; Van Holde, K.E.; Ahern, K. G. (2000). Biochemistry, 3rd edition. Addison Wesley Longman, Inc.
Snustad, Peter, Simons, Michael. (2011). Principles of Genetics, 6th ed. Wiley.