Introduction
Bioinformatics as defined by Merriam-Webster dictionary is the collection, classification, storage, and analysis of biochemical and biological information using computers especially as applied to molecular genetics and genomics. Besides innovations in hardware architecture, networking innovations can also be implemented in order to handle bioinformatics searching.
BLAST stands for ‘Basic Local Alignment Search Tool’, a tool designed by the National Center for Biotechnology Information (NCBI) which was implemented to assist scientists with the analysis of DNA and protein sequences (Chattopadhyay A. et.al.). A public version of this tool is available for use via the web or as a downloadable. The high demand, extensive availability, and overall popularity of BLAST have caused inconsistent performance by its users.
In an effort to improve the performance, students from the Computer Science Bioinformatics Group at the University of South Dakota assembled a BLAST cluster from surplus desktop PCs, which were used to implement a parallel version of the BLAST tool on a Linux cluster. This provided a noticeable searching performance improvement for a much smaller group of researchers. BLAST can be installed on a Linux machine with an Apache Web server running on it. When users submit queries to BLAST a Perl script is invoked which creates the appropriate job to run based upon the parameters entered into the query form. This job is submitted to Open PBS to be executed, based upon the scheduling policy utilized and node availability.
Bioinformatics has become a major field of study due to its importance to the scientific community. Scientists rely on this discipline to provide an efficient and reliable method for the analysis of DNA and protein sequences. The major search tool that’s utilized in this area is called BLAST, for which many other software and hardware solutions have been developed to better utilize and optimize its functionality.
Some example solutions available for performing optimized Bioinformatics database searching over the internet include options from CLC Bio, including their Bioinformatics Cell and Cube, and the Systolic Accelerator for Molecular Biological Applications (SAMBA) each of which makes optimal use of the Smith-Waterman algorithm, and the European Molecular Biology Open Software Suite (EMBOSS). The EMBOSS utilities are a free Open Source software package that was developed around the special needs of the molecular biologist since it is designed to handle and format a variety of data as well as allowing web-based sequence data retrieval. Other options, which have been explored in the thin field, include storage area networks, grid computing, multi-core CPU’s, and even clusters of outdated used computers.
BLAST Sequence Analysis & Search Tool
The unprecedented development in the field of modern-day biological research has witnessed an acceleration in intensive biological sequence comparison and analysis tasks. The comparison of nucleotide or protein sequences from the same or different organisms is a very essential requirement in modern-day molecular biology. (Madden T., 2003). Identification and tracing of the function of newly sequenced genes, prediction of genetic developments, and exploration of evolutionary genetic relationships and functions are no more miracles under the spell of biological sequence comparison and analysis.
The whole genomes can be sequenced now and the similarities between sequences can be searched and ascertained which can finally help predict the function of protein-coding and transcription-regulation regions in genomic DNA (Korf et.al., 2006). One such searching tool most frequently used by biomedical researchers is the Basic Local Alignment Search Tool (BLAST) which can be adapted for use with different query sequences against different databases (Altschul et.al., 1990).
This user-friendly tool was developed by the NCBI (National Center for Biotechnology Information) and was initially released in 1990. The NCBI is a wing of the National Institute of Health located in Maryland and it was founded in 1988 through legislation. This institute operates as a national resource center for molecular biology whose primary functions include- the creation of public databases, conducting research in computational biology, developing genome data analysis tools and dissemination of biomedical information. The NCBI is the authorized custodian of the genome sequence databank, biomedical research repositories and articles and other biotechnology related information.
This institute stores the genome sequence data in the well-known computer database called the GenBank whereas other research repositories and articles are staked in two net-based information libraries well known as PubMed Central and Pubmed (see Madden T., 2003). There is one popular search engine called the Entrez search engine which can facilitate in searching information and databases more efficiently. This tool can help search out the high scoring sequence alignments between the query sequence and sequences in the database adopting a heuristic approach that is in-line with the Smith-Waterman algorithm (See, Robert R. et.al., 2006).
Large genomic databases like the GenBank cannot be searched efficiently using the Smith-Waterman approach as it lacks the speed for searching such large databases. However, this can be accomplished by using the BLAST algorithm which can be 50 times faster than the Smith-Waterman approach subject to using a heuristic approach. BLAST programmes are well known for speed, accuracy and continuous innovations.
BLAST- Functionality & Execution
BLAST standouts to be the most popular and most frequently used search engine in the domains of modern-day bioinformatics, Its simple application protocols and adaptability to fit into a wide range of databases make it an automatic choice for the biotechnology professionals (NCBI, 2003). This tool has undergone continuous development since its release in 1990 and gradually it emerged as the most widely used bioinformatics tool for sequence searching. Using a query sequence and a sequence database as its inputs, BLAST searches all entities in the database for those with high-scoring gapped alignment to the query, where the deletion, insertion and substitution are allowed in sequence comparison and the alignment scores are determined statistically and heuristically based on expert specified scoring matrix (Archuleta J. et.al., 2007).
BLAST performs local alignments only and it uses a heuristic programme as the algorithm that enables it to take a shortcut route to performing the search operation faster. Through local alignment, one mRNA can be aligned with a genomic DNA required in case of genome assembly and analysis. For executing a search operation, the user needs to provide certain input information like- sequences, preferred database, word size, expect value and others.
Once the inputs are provided, the BLAST starts to construct a look-up table of all the words placing them in the short forms or short sub-sequences along with the similar or neighboring words in the query sequence (See, Lipman & Pearson, 1985; Lin Qi, 2005). The sequence database is then scanned for the specific search words which upon identification of a match are used to initiate gap-free and gapped extensions of that word. It may be noted that the BLAST does not search the database files (GenBank files) straightway on its own instead, sequences are made into the BLAST database.
Thus each entry is divided forming two files in effect- one having only the header information and the other having the sequence information only and these become the data the algorithm uses finally. When the BLAST runs in the stand-alone mode then the data file may include local data, private data and the NCBI databases which can be easily downloaded. There may be a combination of the local and private data as well. Following the look-up operation, the algorithm assembles the best alignment for each of the query-sequence pair and passes this information to a SeqAlign data structure (ASN.1) as shown in Figure-1 below.

After finding a similar sequence to the query, the BLAST proceeds to verify whether the alignment is good and whether it is enough to establish a possible biological relationship. The BLAST uses statistical procedures to produce a bit score and exped value (E- value) for each alignment pair. The bitscore gives an indication as to how good the alignment is. The higher the score, the better the alignment (Lipman & Pearson, 1985; Lin Qi, 2005).
The bit scores from different statistical alignments derived using different scoring matrices can also be compared through normalization. Further, the E-value represents the statistical significance of a given pair-wise alignment, size of the database used and the scoring system adopted. A hit with a lower E- value indicates that the hit is more significant. For example, when the E-value of a sequence alignment is 0.05 then this similarity has a 5 in 100 (i.e., 1 in 20) chance of occurring alone by chance. Statistically, this might be a good figure but biologically it may not hold good and this is why sequence analysis need to determine the ‘biological significance’ (Casey R.M., 2005).
The complete BLAST search tool is a package of five programmes that includes- blastn, blastp, blastx, tblastn and tblastx. blastn and blastp. These five programmes help to perform a nucleotide or peptide sequence search in a sequence database. Notably, blastx and tblastn can perform a sequence search of one type with a database of the other type. This is possible as nucleotide and peptide sequences can be translated into each other in living cells. This means a nucleotide sequence can be translated into a peptide sequence and then compared with a database of peptide sequences, and vice versa. Interestingly, the tblastx stands out to be different from all the four programs above.
Tblastx initiates six-frame translations on both the query and the database entities before the commencement of the actual comparison task. All these five programmes are integrated into a single interface and offered as a single unit. NCBI has these five programmes integrated into a single interface commonly known as blastall which makes it easy for a user to access all the five different comparison programmes (Mount D.W., 2004).
BLAST happens to be the most widely used bioinformatics tool for comparing unknown genomic or aminoacidic biological sequences (query) against that of any known sequences that is already included in the referred database. This tool approximately relates to the Smith Waterman algorithm specifically for local sequence alignment resulting in a speed increase of 10100x. This increase in speed comes at the expense of some sensitivity of the BLAST tool. There are different ways through which the problem of BLAST speed can be addressed (Archuleta J. et.al., 2007). There already exist some emerging computer hardware and software solutions for tackling this problem.
BLAST- Workflow & Clusters
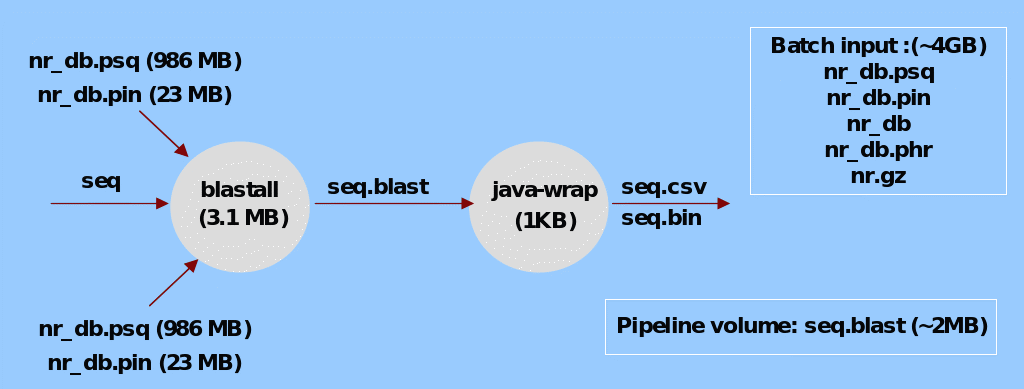
BLAST is a sequence alignment workflow, which given a protein sequence “seq”, can check a database of known proteins for any similarities. Proteins with similar sequences are expected to have similar properties. Finally, the results are converted into CSV and binary format for future using Javawrap (See, Aleksander et.al., 2007).
The Basic Local Alignment Search Tool (BLAST) developed by The National Center for Biotechnology Information (NCBI) is the most widely used bioinformatics sequence searching and analysis tool. Having a huge base of users connecting through its website and being used indiscriminately, the BLAST runs the risk of showing up inconsistent performance sooner or later. To do away with the risks of inconsistent performance, the University of South Dakota (USD) went on to implement a parallel version of the BLAST tool using a Linux cluster which has an interface consisting of a combination of freely available software. Using old or surplus PCs and catering to a small group of researchers, this BLAST cluster has been found to improve the searching process (Mark Schreiber, 2007).
Here the Open Source Cluster Application Resources (OSCAR) developed by the Open Cluster Group has been implemented. This OSCAR improves cluster computing by providing all the necessary software to create a Linux cluster that comes bundled in one package. This helps automate the installation, maintenance and even the use of cluster software. The BLAST cluster used WWW BLAST having web interface which was found to enhance the overall usability of the cluster.
However, improvement in overall performance was observed when mpiBLAST was used (Bowers & Ludascher, 2005). This mpiBLAST can execute queries in parallel thereby improving the BLAST performance. This tool is based on the Message Passing Interface (MPI) which is a common software tool commonly used for developing parallel programs. The mpiBLAST provides all software necessary for parallel BLAST queries.
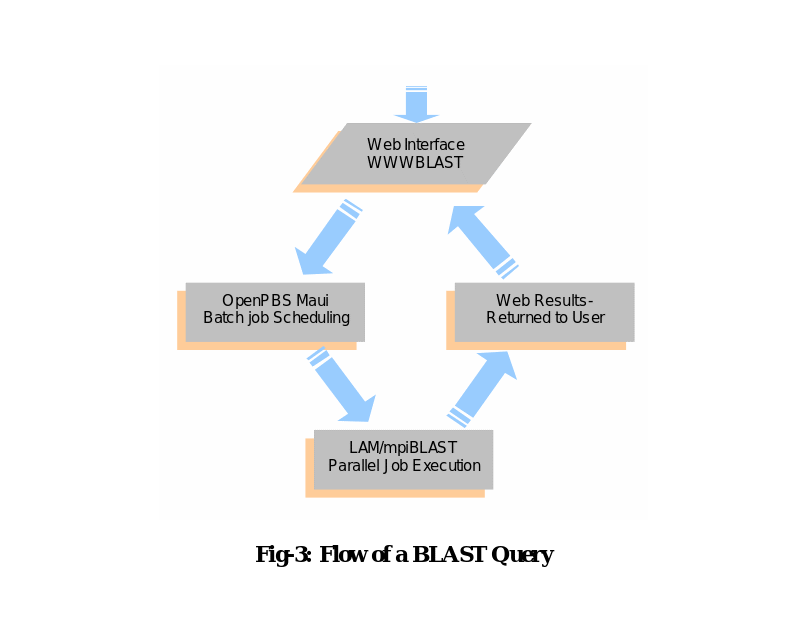
The BLAST search begins with a web-based query form. Batch processing as well as job scheduling are not supported by WWWBLAST but these jobs are taken care of by the OpenPBS and Maui which are included in the OSCAR software suite. The OpenPBS is a flexible batch queuing system originally developed for NASA and Maui is the add-on which extends the capabilities of OpenPBS by allowing more extensive job control and scheduling policies (Oinn, et.al., 2005).
Following the submission of a query by the user, a Perl script appears which creates a unique job taking parameters from the query form. The OpenPBS programme is responsible for determining the availability of node and then execution of the job. After this starts a local area multicomputer (LAM) software which has a user-level daemon-based run-time environment. The LAM is included in the OSCAR installation package and this provides most of the services required by the MPI programme. One mpirun command executes the query and gathers results for each node and the same is actually executed by the OpenPBS (See, Mount D.W., 2004). Finally, all the results are transmitted back to the browser by the WWWBLAST and the same are presented to the users.
Figure-3 above shows the different stages a query passes through once it is submitted through the web. Getting a query from the web, WWWBLAST submits a job to OpenPBS. The OpenPBS starts the job with mpirun and finally the results are formatted by the WWW BLAST. For performing parallel BLAST searches, implementation of the cluster technology becomes a necessity. A parallel BLAST cluster starts working after some initial configuration which is in contrast to other common tools that works with default installations.
Setting up of the cluster is facilitated by an application called the ‘OSCAR’ (Open Source Cluster Application Resources) which is equipped with all the necessary programmes and protocols for putting the cluster into work ( See, Zhang H., 2003). A brief gist of the different platforms and components producing the parallel BLAST cluster is as below:
- OSCAR- stands for Open Source Cluster Application Resources that include the best known methods for building, programming, and using clusters. There is no requirement for any downloading once OSCAR is available. This is because, the OSCAR software includes everything needed to install, build, maintain, and use a modest sized Linux cluster.
- WWWBlast- this is almost like the regular BLAST which implements a web-based interface along with other programs for web based queries. It also provides web-interface to execute BLAST queries against cluster. Here, the search page in HTML format is forwarded to the blast.cgi which initiates the WWWBlastwrap.pl, which is nothing but a Perl script linking on to the openPBS (Leong Philip, 2003). The command line arguments required for the mpiBLAST query exist with the WWWBlastwrap.pl.
- MpiBLAST- is the open source parallelization of BLAST that partitions the BLAST database according to nodes available (command line argument). This MpiBLAST can help process the BLAST queries on multiple nodes simultaneously.
- LAM/MPI- is the local area multicomputer that is for open source implementation of MPI. This application also provides the standard MPI API along with debugging and monitoring tools. Running on a wide variety of Unix platforms, this tool has more control over MPI jobs.
- OpenPBS/Maui- this is a NASA developed batch queuing system which is flexible and capable of initiating and scheduling execution of batch jobs. The capability of this OpenPBS can be extended by better and extensive job control and scheduling which is possible by using Maui (Ligon W.B., 2002). Maui queries the PBS Server and PBS MOM’s to obtain up to date job and node information, PBS manages the job queue and the compute resources. Having all these components make it possible for the cluster to search up-to-date database with fewer concurrent users and help achieve better overall throughput times. This type of cluster is observed to consume less time to complete a query.
Smith-Waterman Algorithm
One of the most frequently used algorithms for performing local sequence alignment is the Smith-Waterman algorithm. Sequence alignment calls for determining similar regions between two nucleotide or protein sequences. This algorithm does not consider the total sequence rather it compares segments of all possible lengths and finally optimizes the similarity measure.
The main aim of the Smith-Waterman Algorithm is to compute the edit distance between two sequences (either DNA or protein) following a dynamic programming approach (Smith & Waterman, 1981) and this is accomplished by creating a special matrix for the cells to indicate the cost to change a subsequence of one to the subsequence of the other. This algorithm progresses accounting for the edit distances of the subsequences thereby turning out to be an efficient way of comparing sequences. The original algorithm, as conceptualized by Smith and Waterman, has some inherent complexities that can make it difficult to achieve the goal.
The core function of this algorithm is database similarity searching between a nucleotide or protein query sequence of interest and sequences in a database. There are multiple reasons for taking up this searching activity some of which include (See, Pearson, 1991)-
- Identify conserved domains in nucleotide or protein sequences of interest to predict functions of new and uncharacterized sequences
- Compare known sequences and identify homology between these sequences
- Search sequences in a database for motifs or patterns similar to motifs or patterns in the sequence of interest
- Search for a nucleotide sequence matching a protein sequence of interest as well as the other way around
- Compare sequences within taxonomic groups
Identification of the good alignments between sequences which are just distantly related may be difficult. There exist low similarity zones which may pose difficulty in identifying the good alignments and thus, concentrating on local alignment searching instead of the global ones is the most prudent option. Targeting the local alignment searches provides the option to analyze for homology even between sequences containing regions of genetic variations (Smith & Waterman, 1981). This algorithm searches for homology by comparing sequences. Comparing sequences using local alignments prioritize estimation of the total number of alignments and identification of the best alignments which finally influence the reliability and relevance of the data obtained (Pearson & Lipman, 1988). The Smith-Waterman algorithm follows the same route.
The Smith-Waterman algorithm uses dynamic programming. Dynamic programming is a technique which is used for dividing problems into sub-problems and solving these sub-problems before putting the solutions to each small piece of the problem together for a complete solution covering the entire problem (See, Altschul et.al., 1997). Thus, the Smith-Waterman algorithm identifies the optimal local alignments following the dynamic programming approach. On its way to finding the optimal local alignments, this algorithm scans through alignments of any possible length that starts and terminates at any position in the two sequences under comparison.
The basis of a Smith-Waterman search is the comparison of two sequences A = (a1a2a3…an) and B = (b1b2b3…bn).

The Smith-Waterman algorithm uses individual pair-wise comparisons between characters as:
In the above equation, Hij is the maximum similarity of two segments ending in ai and bj respectively. The similarity of residues ai and bj is represented by a weight matrix under the consideration of either substitution or insertion/deletion. The first term considers an extension of the alignment by extending the two sequences compared by one residue each. The second and third term handle an extension of the alignment by inserting a gap of length k into sequence A or sequence B, respectively. Finally, the fourth term ignores a possible negative alignment score by placing a zero in the recursion. The preceding calculations runs neutral keeping the allowance of the similarity score zero in the expression for Hij signifying that a local alignment can restart at any position in case of a character-to-character comparison (Smith & Waterman, 1981).

Each of the residues for comparison between the two sequences is assigned a score by the algorithm. The comparison of each pair of characters is weighted into a matrix considering every possible path calculated for a given cell. As is the case with a typical matrix cell, the score of the optimal alignment terminating at the coordinates is represented by the value of the matrix cell. This matrix identifies the highest scoring alignment as the optimal alignment as shown in Figure-4 above.
The highest scoring matrix cell considered for arriving at the optimal local alignment stands as the starting point for the algorithm. The path of the highest matrix cell leading to the optimal local alignment is then tracked back through the array until a zero scoring cell is found. The score in each cell reflects the maximum possible score for an alignment of any length terminating at the coordinates of this specific cell (Smith & Waterman, 1981).
Example

The Smith-Waterman algorithm can be exemplified by the comparison of two sequences:
Sequence A: CAGCCUCGCUUAG.
Sequence B: AAUGCCAUUGACGG.
Hardware based Bioinformatics optimization solutions
The growth rate of the biological sequence data that needs to be analyzed is growing exponentially thereby stamping the need for augmentation of computer infrastructures and the urgency for higher throughput sequence analysis outpacing the standard rate of improvement in underlying computing infrastructure. In case of higher throughput sequence analysis, the choice lies between special-purpose dedicated hardware or commodity cluster computing. This section runs through some innovative options in this field and explores the pros and cons of each option finally outlining a general framework for evaluating alternatives.

CLC Bioinformatics Cube
CLC Cube is a small hardware device used for high performance database searches that are designed to perform at very high speeds without compromising the quality of the search results (CLC Bio., 2006). This device is quite different from the well-known BLAST searches that focus more on speed then the quality of search results. CLC Bioinformatics Cube is built on cutting edge technology and innovative design, which redefines the utility of hardware-accelerated bioinformatics.
This Cube provides a powerful, flexible and integrated hardware accelerated bioinformatics solution thereby doing away with the problems of high-performance computing. Access to this device is also easy using FPGA chips through USB port facilitated by GUI (graphical user interface). This comes as a 12 X 12 X 12 cm steel framed cube having two FPGA chips (Field Programmable Gate array) mounted on boards connected to a computer through a USB port. The device has external memory and is designed to have multiple features like- it should be fast/have high sped, should be user-friendly, should be compatible for integration with existing IT facilities, etc. (CLC Bio. White paper, 2006).
The CLC Cube has FPGA chips that help perform calculations in a highly parallelized environment and also make it reconfigurable thereby rendering a huge advantage compared to normal computers. However, there is a disadvantage associated to the FPGA chips that emerges from the fact that programming these chips is difficult, which may make implementation of algorithm a very time-consuming affair. Its software happens to be the important component adding towards its user-friendliness.
There are two options available for accessing this cube- either via the command line, or through the graphical user interface of CLC bio’s other bioinformatics software applications like CLC Gene Workbench, CLC Protein Workbench, and CLC Combined Workbench. The most compatible access option is the command line version which being based on NCBI BLAST, has similar input and output formats (Haibe & Bontempi, 2006).
Having the innovative FPGA-based chip design and running the Smith-Waterman algorithm enable this cube to achieve a performance per accelerator unit of at least – 50 times that of a fast 3 GHz/4 GB RAM desktop computer. The cube can be easily operated from any CLC workbenches like- CLC Free Workbench, CLC Protein Workbench, CLC Gene Workbench – or the CLC Combined Workbench that aggregates all of the features contained within the multiple workbenches (CLC Bio. White paper, 2006). The other option at hand is using a command-line interface which can be used anywhere at any time just by plugging two cables to a small external box – one cable for power and one for a USB connection. Therefore, the main points which come up about the CLC cube are-
- BLAST is the better in the explorative phases of the research, and Smith Waterman is the better in the late stages of the research, where absolutely correct results are more important than estimates.
- Given a query sequence data with too many of inherent errors, BLAST searches would often be so imprecise that the results are wrong or misleading.
This cube can significantly alter performance by allowing database searches at a high quality level within a reasonable span of time.
Workflow & Databases
A Cube Smith Waterman BLAST search has the following workflow (See CLC Bio. White paper, 2006):
- The user defines the query sequence dataset, the database to be searched, the e-value, and the scoring matrix to be used.
- Either a command line interface just like the BLAST command line interface of the NCBI or a graphical user interface of the CLC Bio software application starts the Smith Waterman BLAST. Good results can be obtained using any of these two options, which can be shown, graphically in a table or as text like the traditional BLAST output.
- The final output shows a number of alignments between the query sequences and the database sequences in pair formation and the e-value signifies the number of aligned sequences.
At lower e-values, the cube is found to achieve better performance and the same is possible as the alignment part of the Smith Waterman BLAST is made in the software, which by nature is not sped up in the Cube.
Database searches & Benchmarking

There are different types of database searches as listed in Table-1 below. The Smith Waterman based BLAST searches can be run on this cube.
Simple comparisons have been done taking these platforms into consideration- CLC Bioinformatics Cube, clcblast_0.2.0 and a DELL Desktop with a 3.0 GHz Pentium 4 CPU, and 2 GB of RAM. For this, the Smith Waterman BLASTn was tested on a dataset consisting of all horse EST sequences in GenBank (36,914 sequences with a total number of residues of 19,762,562). In case of the BLASTp, the test dataset included 50,000 protein sequences randomly chosen from the SwissProt (with a total of 18,396,764 residues).
The execution time represented as the ‘speed-up’ signifies the execution time of the algorithm without using the Cube as against the execution time of the algorithm when the Cube is plugged in. A similar approach has been followed earlier as referred in (CLC Bio. White paper, 2006).
Smith Waterman BLASTn- Benchmark
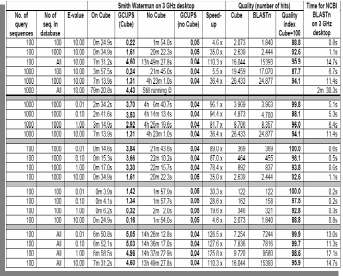
As reflected in Table-3 below, the 3 GHz Desktop is quite slower compared to the speed of the cube when considered for small amounts of query sequences and small datasets and the speed increases further in case of larger amounts of query sequences especially on larger datasets.

The cube can achieve a speed 4-8 times faster then the 3 GHz Desktop in case of small quantity of sequences and small datasets. As reflected in Table-2, the cube speed is 35 to 128 times faster than the 3 GHz Desktop in case of databases above 500,000 nucleotides. When the e-values are at 1 or below, the ‘speed-up’ is always found to be above 78 times. The cube runs at a speed of up to 5 Giga Cell Updates Per Second (GCUPS) as against speed of up to 0.05 GCUPS that the 3GHz Desktop achieves. However, in case of small databases, the ‘speed-up’ is low at higher e-values, which is attributed to the time overhead incurred by the parts of the searches performed in the software (Haibe & Bontempi, 2006).
Smith Waterman BLASTp- Benchmarks
Given the fact that there are only 4 different nucleotides but 20 different amino acids, a search of the amino acids naturally finds the cube speed reduced compared to that of the nucleotide search. As shown in Table-5, the cube speed in connection with lesser number of sequences and smaller datasets is about 3 to 9 times faster then that of the 3 GHz Desktop. The ‘speed-up’ increases in case of higher number of sequences and larger datasets. In case of above 350,000 amino acid databases, the cube achieves a speed of 1.55 GCUPS whereas the 3 GHz Desktop achieves only 0.046 GCUPS. Here, the number of query sequences may influence results but it is believed that the effect will not be very significant (See, Altschul et.al., 1997).


The Smith Waterman versions of tBLASTn, BLASTx, and tBLASTx have almost the same speed-up approximately akin to that of the Smith Waterman version of BLASTp. All these algorithm versions are also capable of performing protein-protein comparisons (See CLC Bio. White paper, 2006).
The FPGA Technology
In case of traditional integrated circuits, there is a large grid having various gates, inverters, etc. connected that finally controls the system and performs the desired functions. The CPUs of computers are akin to the integrated circuits but have enhanced functionalities. The CPUs are required to perform multiple functions such as- adding integer numbers, multiplying real numbers, reading and writing to memory, etc. The CLC Cube uses an innovative technology called the FPGA technology, which houses a large number of gates in an integrated circuit to perform the function of simulating how an integrated circuit works (Smith & Waterman, 1981).
Thus, an integrated circuit when upgraded into a FPGA can simulate the circuit works thereby function like a specially designed chip. One unique advantage in this case is that many circuits can be upgraded and uploaded into FPGA making it possible for the unit to perform multiple functions. However, FPGA programming is not that simple and this cannot be done using the typical gates having two input wires.
The FPGI technology uses lookup tables with four inputs making it more advanced than the normal gates. The outputs of the lookup tables can be integrated by connecting them to the input wires including other tables forming a large network. It is just like the standard integrated circuits and the FPGA chips characteristically have small amount of internal memory, which can be used for remembering various states and for storing some data.
The output from these lookup tables can then be connected to the input wires for other tables in a large network, just like standard integrated circuits. Moreover, the FPGA chips have a small amount of internal memory, which can be used for remembering various states and for storing some data. It may be noted that an FPGA does not have a fixed clock speed. The specific circuit design uploaded to the FPGA determines the clock speed in reality. FPGA has programmable clock speed, which is adjusted to suit the specific calculations to be performed.
The CLC Cube has two FPGA chips mounted on boards with external memory and adding one extra external memory to the boards facilitates storing of large amounts of data. Following this option helps to wipe off the disadvantage associated to the commonly used limited internal memory of a chip. One USB connection connects the boards to the computer. The current versions of USB connections are found to have very high capacity however, the very high computing power of the FPGAs pose a challenge to design the algorithms fitting the USB bandwidth (CLC Bio. White paper, 2006).
SAMBA- Algorithm & Architecture
SAMBA has been designed at IRISA to cater to certain specialized applications like biological sequence comparison, image compression, data compression, coding and cryptography (Lavenier Dominique, 1996). It is a full custom systolic array based hardware accelerator, which is capable of carrying out comparisons of biological sequences with good success. This hardware accelerator implements a parameterized version of the Smith and Waterman algorithm allowing the computation of local or global alignments with or without gap penalty (Smith & Waterman, 1981). The speed-up provided by Samba over standard workstations ranges from 50 to 500, depending on the application. Basically it is a hardware accelerator designed for speeding up the algorithms involved in biological sequence comparison (Guerdoux & Lavenier, 1995).
SAMBA Algorithm
The main algorithm SAMBA runs on belongs to the dynamic programming class and the recurrence equation comes from the Smith and Waterman algorithm. However, the algorithms have been customized and parametrized to cover a larger domain. In SAMBA, a similarity matrix is calculated recursively using the following equation (See, Lavenier Dominique, 1996):

and the initializations given by:
H(i,0) = E(i,0) = hi(i) H(0,j) = F(0,j) = vi(j)
alpha, beta, delta, hi and vi are the parameters for tuning the algorithm to local or global search, with or without gap penalty.

The process of sequence comparison on a linear systolic array proceeds with one query sequence stored into the array comprising one character per processor followed by other sequence flowing from the left to right through the array. Each processor is engaged in computation of one elementary matrix during each systolic step. The result appears on the rightmost processor when the last character of the flowing sequence is output.
Compared to a sequential machine the speed-up for computing one query sequence of size N against a database of M residues is given by (Lavenier, 1993):
N x M
——— = N (considering that M >> N)
N + M – 1
N = size of the query sequence (number of processors)
M = size of the database
SAMBA Architecture

The SAMBA workstation houses a local disk, a systolic array with 128 VLSI full custom processors and a FPGA-Memory interface. The FPGA-memory fills the gap between a complete hardwired array of processors and a programmable Von Neuman machine (Figure-8) (Lavenier Dominique, 1996).
One array with a set of 32 full-custom identical chips housing 4 processors each leads to a 128-processor array and the computational power generated by the chip is about 40 millions matrix cells per second. This power of computation is quite sufficient to enable the array to clock a peak performance of 1.28 billions matrix cells per second.
Basically, SAMBA is a hardware accelerator having a library of high level comparison C-procedures which makes possible the choice of pre-defined programs, such as the classical programs already developed for bank-scanning, or user-defined programs tuned to a specific application. It is controlled by a few procedure calls inside a normal C-program thereby not mandating the user to have specific knowledge of the structure of the accelerator and how it works (See, Pearson, 1991).
Grid Technology
Grid technology has emerged as the most viable option for integrating large-scale, distributed and heterogeneous resources and off late, it is making substantial in-roads into the field of bioinformatics. In the domains of bioinformatics, grids form the technology backbone, which facilitate and enable sharing of bioinformatics data from different sites and databases scattered throughout the world. Unlike the centralized database system, grid technology helps in sharing databases, which are geographically distributed and thus it contributes towards eliminating the scopes for single-point failure of dataflow commonly encountered in centralized databases (Vazhkudai et.al., 2001).
Under the grid environment, any new set of data like experiment results can be easily stored on a local system and shared with other systems distributed throughout the world. Moreover, the users can access and work with the data at their ends without even knowing the actual location of the target data or information. The grid system handles data coming from various sources and different locations and this arrangement demands the grid system to have compatible computing infrastructures ensuring free flow of data. Thus, this diverse configuration of target data influences the design of grid-infrastructures thereby calling on the middleware layer to act as the interface for transforming the various data sets into a unique standard format (See, Wasinee et.al., 2002).
The grid infrastructure is best suited for parallel and distributed applications and above this, grids also provide seamless access to the data repository. The grid resources can be used to perform tasks like dynamic simulation, network modeling and other computationally intensive tasks. Moreover, the grid network is a confluence of computer-related resources and the system is bestowed with geographically distributed computing power that can enormously aid in the data-sharing task, which is characteristically the most important function of any grid system (Vazhkudai et.al., 2001).
In case of a bioinformatics grid setup, it is important to manage and maintain the distributed computing components in the bioinformatics application for ensuring efficient and barrier free access to the distributed data coming from various locations. Accomplishing this requires a software system design and creation of which happen to be very challenging tasks. Besides managing distributed computing components, this system should be able to minimize the communication costs by identifying the best option for computing the datasets.
Here the software needs to select one of the two computing options- computing by sending data to a dedicated system or computing by using a computation object at the point of the target database (Wasinee et.al., 2002). Despite being a complex system, superlative computation power, huge volume of data handling capacity and the extended geographical coverage of the grid system make it the most preferred option in the modern biology and bioinformatics domain.

Figure-9 above depicts the architecture of a multi-institutionally operated, distributed database dependent and heterogeneous computing environment exposed grid system. There is a search engine running underneath the grid data system, which feeds the locations of the target data. After identifying the data locations, data selection and processing is undertaken inline with the specifications of the preferred computing method. Sometimes the computing task demands extra inputs which, if available locally, can be provided directly.
In case the extra inputs are not found locally then the process can be recursively applied. The above shown architecture is for generic grid data services with a grid security system which must be implemented as a gateway for every grid site thereby providing uniform access to heterogeneous storage systems including vendor’s databases, XML files and proprietary bioinformatics files as well. There is a storage system application-programming interface (API) that facilitates uniform access to different storage systems (Kola et.al., 2005). For using this system, the users are not required to know about the operations of each storage system.
There is an open standard protocol called the LDAP on top of which the meta-data catalog is implemented and this catalog stores all storage system information. The application specific behaviours such as data access policy in the cache management service are present in the top layer of this architecture. Such an arrangement makes reusability of the basic grid data architecture possible and at the same time provides flexibility to the users to tailor their applications to achieve high performance (See, Wasinee et.al., 2002).
Bioinformatics Sequence Searching Options- Evaluation & Analysis
Tools & Methodology
In the field of Bioinformatics, sequence similarity searching and multiple alignment applications are the preferred approaches followed particularly in case of large databases. Searching large databases generally takes more execution time which is higher and impracticable for taking-up a simulator-based study. The study at hand aims at collecting data with reasonable input sizes covering the complete execution phase of the bioinformatics applications.
Taking this into account, it has been planned to use the hardware performance counters. The hardware counters used here are built into modern microprocessors instead of a simulator. Many special purpose counters are available today that can be used to count different events like- cache misses, branch mispredictions, and others that are useful measures of application performance (See, Kursad et.al., 2005). Their limited number happens to be the only problem or drawback associated to hardware performance counters. This study uses only 2 counters on Intel Pentium-III CPU. This drawback has been countered here by multiplexing.
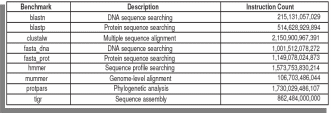
The multiplexing uses time-sharing to use the counters to measure different events at different time slices, and extrapolates the result and this method is found to give reasonably accurate measurements. The PAPI hardware performance counter access library has been employed here that uses the perfctr Linux kernel patch for counter multiplexing (Brown S. et.al., 2000). Data collection and analysis have been carried out using the PerfSuite utilities. This set-up helped to run unmodified BioBench applications with large input sizes characteristic of their typical use. The entire workloads took up to 2.1 trillion instructions and the number of instructions for each BioBench suite is given in Table-6 below.
Benchmark Characteristics & Evaluation
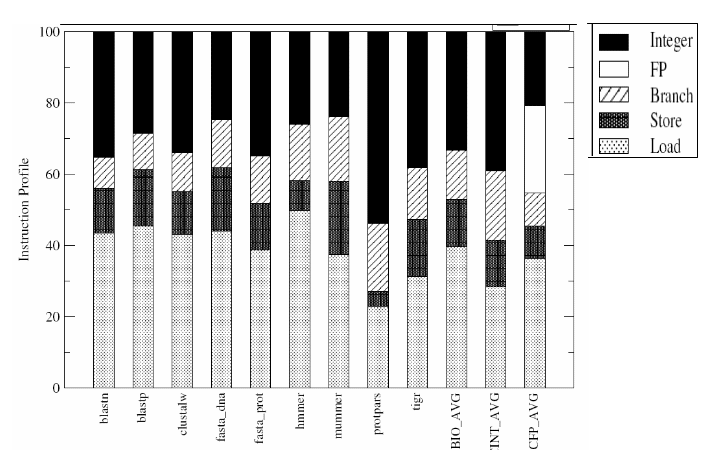
Aseries of data have been collected for characterizing the BioBench suite. The data collected include- data on instruction profiles, basic block lengths, IPC, L1 and L2 data cache miss rates, and branch prediction accuracy. The same set of data was collected for the SPEC 2000 benchmarks as well for comparison. In the first phase, the hardware performance counters were used to provide a count of instructions belonging to different major instruction classes in the X86 architecture. The instruction profiles for BioBench applications are given in Figure-10.
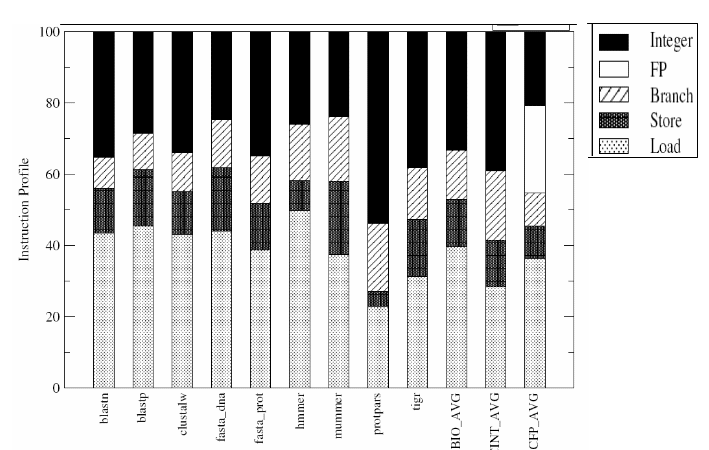
From the figure it can be observed that the floating-point operation content of almost all BioBench applications are negligible and this is in-line with the understanding that bioinformatics applications are inherently different from mainstream scientific codes due to the representation of data they operate on. It can be observed that none of the BioBench workloads have a floating-point instruction content of over 0.09 % of the total instructions executed.
Further, the share of load instructions in BioBench applications does not match with that of the SPEC integer benchmarks which implies that the per datum computation amount is relatively small. The small amount of computation happens to be a typical characteristic of the search algorithms. It can be seen that the Protpars is the benchmark with the smallest input file size and this benchmark also differs from the rest of BioBench components having larger share of integer ALU instructions accounting for more than half of the instruction count. BioBench has higher share of load/store operations which clearly suggests that bioinformatics applications are likely to benefit from future architectures with higher memory bandwidth and prefetching.

The IPC numbers for the applications in the BioBench suite have been shown in Figure-11 above. The BioBench benchmarks are found to have significantly higher average IPC and this indicates that the BioBench applications have higher levels of ‘Instruction Level Parallelism’ (ILP) compared to that of the SPEC INT and FP benchmarks. This finding augers well with the previous finding of almost negligible floating point content in BioBench which conclusively suggests that the bioinformatics applications will benefit greatly from wider superscalars of the future with highly optimized fast integer cores. It may also be seen that there is considerable variations in the IPC values for all the individual applications in BioBench system.
The basic block length for BioBench applications has been depicted in Figure-12 below. The figure shows that the average basic block length of BioBench applications lies roughly between those of the SPEC INT and SPEC FP averages and the all individual BioBench benchmarks are having higher basic block lengths compared to the SPEC INT average. This higher block length signifies that bioinformatics applications are closer to scientific workloads than in terms of the distribution of control transfer instructions.

As shown in Figure-13, the branch prediction accuracy for the BioBench benchmarks is relatively lower than that for the SPEC benchmarks. The difference between the branch prediction accuracies in case of the two benchmarks do not appear to be very significant given the very high prediction accuracy available with modern branch predictors.


Figures-14 and 15 present the data cache miss rates and highlight differences in memory usage patterns of different BioBench components.

Figure-14 indicates that the mummer and tigr have higher L1 data cache miss rates compared to the rest of the applications in BioBench benchmarks and the same is reflected in their L2 data cache miss behaviours (Figure-15). The data cache miss rates of the multiple alignment component clustalw shows very low L1 and L2 whereas the clustalw shows high IPC and fairly high average basic block length along with its low memory footprint.
This evaluation authenticates the common belief that bioinformatics applications have characteristics that distinguish them from traditional scientific computing applications characterized by SPEC FP benchmarks. There is no significant floating-point instructions observed in case of the bioinformatics applications evaluated in this study. Having higher ILP and having basic block lengths closer to the SPEC FP benchmarks compared to that of the SPEC INT counterpart indicate uniform regularity in distribution of branches. Therefore, it can be said that bioinformatics applications stand to benefit from future architectural features such as increased memory bandwidth, memory prefetching and wider superscalars to exploit their high ILP.
Conclusions
This paper confirms the fact that there exists a series of bioinformatics tools and techniques but identification of the best option is never easy. This study identifies some issues which are-
- Biological data analysis and management are still quite difficult jobs because of the lack of development and adaptation of optimized and unified data models and query engines.
- Some of the existing bioinformatics ontologies and workflow management systems are simply in the form of Directed Acyclic Graphs (DAGs) and their descriptions are lacking expressiveness in terms of formal logic.
- Lack of open-source standards and tools required for the development of thesaurus and meta-thesaurus services.
- Different problem sizes. While it is easy to normalize performance to, for example, a 10-million residue search, it is more difficult to adjust for different query length.
To sum up, this study finds that bio applications are better for parallel architectures when they are dominated with integer operations and when operations are involved for changing the link trees between on and off is critical.
References
Aleksander Slominski, Dennis Gannon and Geoffrey Fox, ‘Introduction to Workflows and Use of Workflows in Grids and Grid Portals’, Indiana University, 2007.
Archuleta, J.; Tilevich, E.; Wu-chun Feng, ‘A Maintainable Software Architecture for Fast and Modular Bioinformatics Sequence Search, ICSM 2007. IEEE, Issue.
Altschul S.F., Gish W., Miller W. Myers E., ‘ Basic local alignment search tool’, Journal of Molecular Biology, 215:403-410, 1990.
Bowers, S. and Ludascher, B. 2005. Actor-Oriented Design of Scientific Workflows. In 24 th Intl. Conf. on Conceptual Modeling (ER).
Browne S., Dongarra J., Garner N., and Mucci P., ‘A scalable cross-platform infrastructure for application performance tuning using hardware counter’ in Proceedings of the 2000 ACM/IEEE Conference on Supercomputing, 2000.
Casey, RM (2005). “BLAST Sequences Aid in Genomics and Proteomics”. Business Intelligence Network.
Chattopadhyay A. et.al., ‘Design and implementation of a library-based information service in molecular biology and genetics at the University of Pittsburgh’, J Med Libr Assoc., 307-313, E192, 2006.
CLC Bio., CLC Bioinformatics Cube, CLC bio LLC, Cambridge, 2006.
Guerdoux J. & Lavenier D., Systolic filter for fast DNA similarity search’, ASAP’95, Strasbourg, 1995.
Haibe-Kains, Bontempi G., ‘Data Analysis and Modeling Techniques, Bioinformatics Tools for Microarray Analysis’, Microarray Unit, Institut Jules Bordet, Machine Learning Group, Universit´e Libre de Bruxelles, 2006.
Kursad A., Jaleel A., Wu Xue, Manoj Franklin, Bruce Jacob, Tseng Chau-Wen and Yeung Donald, ‘BioBench: A Benchmark Suite of Bioinformatics Applications’, University of Maryland, College Park, 2005.
Korf Ian, Yandell Mark & Bedell Joseph, ‘An Essential Guide to the Basic Local Alignment Search Tool- BLAST’, Oreilly, Cambridge, 2006.
Kola G, Kosar T, Livny M. Faults in Large Distributed Systems and What We Can Do About Them. Proceedings of 11th European Conference on Parallel Processing (Euro-Par 2005).
Lavenier Dominique, SAMBA- Systolic Accelerator for Molecular Biological Applications’, IRISA, Campus universitaire de Beaulieu, RENNES Cedex, France, 1996.
Lavenier D., An Integrated 2D Systolic Array for Spelling Correction & Integration : the VLSI journal, 15:97{111, 1993.
Lin Qi, ‘Parallelisation of the blast algorithm’, Cell Mol Biol Lett. 2005; 10(2):281–5.
Lipman DJ, Pearson WR. (1985) “Rapid and sensitive protein similarity searches”. Science 227(4693): 1435-41.
Leong Philip, ‘Hardware Acceleration for Bioinformatics’, Department of Computer Science and Engineering, The Chinese University of Hong Kong, 2003.
Ligon W. B., ‘Research directions in parallel I/O for clusters’, keynote speech, in Proceedings of 2002 IEEE International Conference on Cluster Computing. 2002.
Madden T., ‘The BLAST Sequence Analysis Tool’, National Center for Biotechnology Information (NCBI), 2003.
Mark Schreiber, Bioinformatics Research Investigator, ‘Bioinformatics workflow management- Thoughts and case studies from industry’, Novarties Institute of Tropical Diseases, WWWFG, 5-7 2007.
Mount D.W., “Bioinformatics: Sequence and Genome Analysis.”. Cold Spring Harbor Press. 2004.
(NCBI)/National Center for Biotechnology Information (NCBI). 2003. Web.
Oinn, T., Greenwood, M., et al. 2005. Taverna: lessons in creating a workflow environment for the life sciences. Concurrency and Computation: Practice and Experience, Vol 18, Issue 10.
Pearson W.R., ‘Searching protein sequence libraries: comparison of the sensitivity and selectivity of the Smith-Waterman and FASTA algorithms’, Genomics, 11, 635-50, 1991.
Pearson WR, Lipman DJ: Improved tools for biological sequence comparison. Proc Natl Acad Sci U S A 1988, 85(8):2444-2448.
Robert R et,al., ‘A Genetic Algorithm-Based, Hybrid Machine Learning Approach to Model Selection’, Journal of Pharmacokinetics and Pharmacodynamics: 196–221.
Smith T. and Waterman M., ‘Identification of common molecular subsequences’, Journal of Molecular Biology, 147:195–197, 1981.
Vazhkudai S., Tuecke S. and Foster I., ‘In Proceedings of the First IEEE/ACM International Conference on Cluster Computing and the Grid (CCGRID 2001)’, pp. 106–113, IEEE Press, (2001).
Wasinee R., Noppadon K., Chularat T. and Royol C., ‘Grid computing and bioinformatics development- A case study on the Oryza sativa (rice) genome’, Pure Appl. Chem., Vol. 74, No. 6, pp. 891–897, 2002, IUPAC.
White paper on CLC Bioinformatics Cube version 0.2.0, CLC Bioinformatics Cube, CLC bio LLC, Cambridge, 2006.
Zhang, H. (2003). Alignment of BLAST high-scoring segment pairs based on the longest increasing subsequence algorithm. Bioinformatics 19, 1391-1396.