Abstract
This is an analytic review of the studies elaborating on the relationship of hyperphosphorylated tau proteins to the development of Alzheimer’s disease and focusing on the antigen capture ELISA specific for p-tau proteins. The aim is to find specific details which would provide an idea of the pathophysiology of Alzheimer’s disease. This illness afflicts the elderly population producing a cognitive decline and dementia making them totally dependent on carers for their upkeep and daily routine functions. Among the many illnesses which have a cognitive impairment, Alzheimer’s is the one with accompanying dementia. Five studies have been reviewed and the relevant details have been analyzed and compared. The antigen capture ELISA was obtained from another study. This paper has brought out significant points and problems which are still hindering the search for possible treatment or prevention of the illness through early detection as Alzheimer’s is believed to begin building its pathological hallmarks about 20-30 years before symptoms set in.
Introduction
Aging has many illnesses and deficiencies which accompany it. The syndrome of mild cognitive impairment is age-related with growing significance in the modern world where the elderly population is “gaining a strong foothold”. It does not have accompanying dementia. However, the people affected will be worried over the potential onset of dementia or Alzheimer’s disease which also begin the same way. Currently, there is no method by which the differentiation can be made. Memory impairment can also be caused by cerebrovascular disease but here risk factors like hypertension would be identified and brain imaging techniques would confirm the diagnosis. Clinically the progress of mild cognitive impairment going onto Alzheimer’s disease (AD) cannot be diagnosed prior to the patient progressing well into the disease. The significance of biomarkers that would detect the impending AD is thus highlighted. Diagnostic accuracy is the challenge so that prospective patients can consume some drugs which can prevent the onset of AD or cure the patient. Research is focusing on biomarkers and their relationship with the CSF proteins.
Neurofibrillation is the neuropathological hallmark of Alzheimer’s disease (Buerger et al, 2006). The cerebrospinal fluid (CSF) of these patients contains hyperphosphorylated tau protein which has been identified as a core biomarker of the illness. Efforts are in progress to identify the biomarkers of Alzheimer’s disease. The proteins in the CSF are being linked to the neuropathological features of the illness (Consensus Report, 1998 cited in Buerger, 2006). The pathophysiology involves the “abnormal hyperphosphorylation of the microtubule-associated protein tau and its incorporation into neurofibrillary changes” (Buerger, 2006). The neurofibrillary changes include neurofibrillary tangles (NFT), dystrophic neurites surrounded by neuritic plaques (NP), and neuropil threads (Buerger, 2006). Research has found that high increases of hyperphosphorylated tau protein (P-tau) have been associated with Alzheimer’s disease (Bureger and Hampel, 2004). Frank et al have already indicated in earlier studies that the P-tau measurement is a core biomarker of the illness AD (2003).
The increase of P-tau231P is related inversely to the cognitive decline in the serial analysis (Hampel et al, 2001 cited in Buerger, 2006). A correlation has been found between P-tau231P and the neurofibrillary tangles in Alzheimer’s disease (Buerger, 2006). The NP is associated with lesser amounts of P-tau231P. The longer disease duration also accounted for more of the three pathological markers in earlier studies (Berg et al, 1998 cited in Buerger, 2006). In Buerger’s study of 2006, disease duration did not seem to change the picture.
The NFTs and the neuritic process are composed of fibrils with P-tau while the plaques contain fibrillous beta-amyloid (Dickenson, 1997 cited in Buerger, 2006). This could be the reason for the relationship between the P-tau231P and the NFTs, while there is a lesser relationship with the plaques. CSF levels of P-tau were found in the neocortical regions of the brain while little was found in the hippocampal region. Hampel et al, 2005, have shown a positive correlation between the high P-tau levels and the rate of hippocampal atrophy.
The pre-clinical phase of the degenerative process in AD lasts for about 20-30 years before the clinical onset of the disease (Davies, 1988 cited in Blennow and Hampel, 2003). During this phase, the plaque and tangle loads increase. The symptoms appear when a threshold is reached. The intensity of the illness is believed to be at one steady level of increase: the degeneration, plaque, and tangle formation do not have periods of differing growth.
The total amount of tau protein indicates the intensity of the neuronal damage and is a state marker (Blennow and Hampel, 2003). The amount in CSF is maximum for severe degenerating diseases like Creutzfeldt-Jakob disease and less in AD (Otto et al, 1997 cited in Blennow and Hampel, 2003). People with minimal or no degeneration have only a normal concentration. The stage marker indicates how much into the disease the patient has progressed. One is the atrophy of the hippocampus. Patients with mild cognitive impairment also have high concentrations of tau protein and hippocampal atrophy but the tau will be greatly increased in relation to the atrophy which is minimal for a 290% increase in the proteins; the degree of atrophy of the hippocampus would be only about 10% (Wolf et al, 2003). The two would be more closely related and the percentage of difference in both will be almost the same in AD (Blennow and Hampel, 2003).
The CSF is close to the extracellular space in the brain. Biochemical changes thereby affect the brain (Blennow and Hampel, 2003). So the CSF is a source of biomarkers in AD and these biomarkers would reflect the pathogenesis occurring like neuronal degeneration, deposition of amyloid-beta peptide in the plaques, and hyperphosphorylation of the tau proteins and the formation of the tangles. Concentrations of the total tau protein in the CSF, the 42 amino acid form of A-beta, and the phosphorylated tau protein (Blennow and Hampel, 2003). The A-beta, which is a cleavage product of the amyloid precursor protein, is a component of the plaques. The phosphorylated tau protein is the newest discovery.
The measurement of the combination of CSF phosphorylated tau protein and Abeta42 has been studied by Maddalena et al (2003) as biochemical markers. 100 consecutive patients were examined. CSF measurements were made on the dementia patients and 31 healthy control participants (Maddalena et al, 2003). The ratio of phosphorylated tau protein to Abeta42 was found to be much increased in patients with AD. The healthy control subjects did not have the increase. The other participants with non AD dementias and other neurological disorders also did not show an increase in the two biochemical markers (Maddalena et al, 2003).
While Buerger (2006) has mentioned that neurofibrillation is the pathological hallmark of AD, Hampel et al (2004) have indicated that “abnormal hyperphosphorylation of the microtubule-associated protein tau and its incorporation into neurofibrillary tangles are major hallmarks of the pathogenesis of Alzheimer’s disease”. Hampel and his colleagues tried to differentiate among patients with AD, non-demented control subjects, and patients with other dementias. The classification of illnesses into Alzheimer’s disease, frontotemporal dementia, and major depression could rely on the results of Hampel et al’s study (2004). Participants included 161 patients of AD, frontotemporal dementia, dementia with Lewey bodies, and vascular dementia. The control was made up of 45 non-demented people. Measures were made of the tau protein phosphorylated at the threonine 231(p-tau 231) level, threonine 181 (p-tau 181) level, and at serine 199 (p-tau199). Different enzyme-linked immunosorbent assays were done for the three levels. P-tau subtype is where the tau was phosphorylated at the level of threonine 231 (P-tau231P) in the diseased brain when compared to controls (Hampel et al, 2004). This subtype is more superior in importance to the subtype at serine 199 (Hampel et al, 2004).
While Buerger has described neurofibrillary changes as the hallmark of the pathology of AD and the hyperphosphorylated tau protein is thought to lead to it, Hampel considers the “abnormal hyperphosphorylation of the microtubule-associated protein tau and its incorporation into neurofibrillary tangles” as the pathophysiological hallmark. Both convey the same information. Buerger intended to investigate how the CSF levels of the P tau protein were related to the neurofibrillary changes in AD while Hampel extended his study to include other dementias and non-demented persons.
Materials and Methods
Buerger’s study used CSF from 26 persons clinically diagnosed with AD. The CSF was stored at -80oC. These patients died within 3 years and findings of the autopsy were collected. Light microscopy, in Bielschowsky silver, impregnated sections in magnification 100 was used to assess the neurofibrillary tangles and the plaques and quantify the hyperphosphorylated P tau protein (Moelsae et al., 1987 cited in Buerger, 2006). The regions selected were the frontal (Brodmann area 9), temporal (22), parietal (39) cortices, and the CA1 region of the hippocampus. (Kohnken et al, 2000, cited in Buerger, 2006). P-tau231P was measured using the CSF and brain homogenates and applying the enzyme-linked immunosorbent assay (ELISA; Applied NeuroSolutions Inc., Vernon Hills, IL, USA). Immunohistochemical methodology of labeling by using the antibody AT8 helped Buerger et al to identify the hyperphosphorylated P-tau protein in AD in the tangle and pre-tangle material. Braak et al have suggested that staging is possible with this technique (2006).
The review by Blennow and Hampel (2004) indicated six different ELISA techniques which showed a high concentration of P-tau protein in the CSF of patients with AD. Sensitivity varied among the various studies selected for different and same ELISAs. As the sensitivity and specificity of the studies cannot be defined, Blennow and Hampel advocate further specific ones (2004).
Maddelena and her colleagues explored the measurements of phosphatase and Abeta 42 in 100 patients who had been diagnosed with dementia and with 31 healthy patients as controls. 51 patients were considered to have AD. Vascular dementia was 8 in number, cerebral amyloid angiopathy 2, Lewy body dementia 2, frontotemporal lobe dementia 3, and Parkinsonism 4 numbers. The CSF of the 100 patients was measured for phosphatase and Abeta 42. A sandwich enzyme-linked immunosorbent assay was adopted.
Hampel (2004) studied the association of p-tau proteins at three amino acid levels: threonine 231, threonine 181, and serine 199. Three different enzyme-linked immunosorbent assays were done. 206 individuals were participants. 108 of these were believed to have AD. CSF p tau231 was used to distinguish AD from other neurological disorders and AD from non AD dementias, especially frontotemporal dementias. Patients with AD had a high level of p-tau231. Hampel’s was the first study using three different assays.
Antigen capture Elisa tests specific for p-tau proteins
Six methods of ELISA have been used to show a high concentration of p-tau proteins in the CSF of AD patients (Blennow and Hampel, 2003). An antigen-capture ELISA test with specificity for p-tau proteins is described by Maddalena and her colleagues in their study (2003). Monoclonal antibody AT270 is specific for tau proteins phosphorylated at threonine 181. This was used in a sensitive sandwich ELISA. Seventy-five μL of CSF samples and Microtiter plates precoated with HT7 (tau epitope 159-163) were incubated together (Maddalena, 2003). The detector antibody was the washed and biotinylated AT270. This was incubated with peroxidase-conjugated streptavidin and then washed once more where a chromogen,
3,5,3’,5’-tetramethylbenzidine, is added. Absorbance was read as 450nm (Maddalena, 2003). “The CSF samples and standards were assayed in duplicates” (Maddalena, 2003). ROC analysis was then done for the CSF ratio of phospho-tau to Abeta42.
Hu et al have described a more detailed antigen capture ELISA for p-tau proteins (2002). The first step involves the coating of a microtiter plate with tau anti-serum 92e (1:2500 dilution) in a proportion of 100 μl/well. This process is done during the night at 4oC. An alkaline solution known as TTBS of Tris-buffered saline (TBS) with pH 8.5 and containing 0.05% Tween 20 is used to wash the plate 5 times. The second step of blocking involves the addition of the blocking solution. This solution is composed of TTBS to which 3% of bovine serum albumin is added (Hu et al, 2002). 150 μl/well of it is added and incubated at 37oC for one hour. Washing is repeated. The third step involves the addition of antigen. This is an overnight process where the plate is incubated at 4oC along with 20 μl of CSF which is diluted to 100 μl in TTBS, 3% bovine serum albumin, and 0,02% NaN3. Then the plate is washed. The fourth stage involves the addition of a reported or primary antibody,100 μl of Tau-1 (1:50,000) or PHF-1 (1:200) to the plate (Hu et al, 2002). The incubator is set at 37°C for 1 hour. The wash completes this step.
The fifth step which involves the addition of the secondary antibody is similar to the 4th step but the solution added is 100 μl of alkaline phosphatase-conjugated goat anti-mouse IgG (1:10,000). Washing is done at the completion of the step. The sixth step involves the initiating reaction. 100 μl of freshly prepared initiation buffer is added to the plate, Incubation is done at 30oC for 45 minutes (Hu et al, 2002). The buffer is 225 mmol/L diethanolamine, pH 9.5, 0.04 mmol/L MgCl2, and 1 μmol/L NADP+. The seventh step begins with the transfer of the initiation mixture into an Eppendorf tube. After the addition of the 100 μl of enzyme-substrate recycling solution, half an hour’s incubation is done at 37oC. The enzyme-substrate solution is 0.1mol/L phosphate buffer, pH 7.4, 6% alcohol, 0.5 U/ml alcohol dehydrogenase, 0.0125 U/ml diaphorase, and 8μ mol/L resazurin. The last step involves the measuring of the fluorescence after stopping the reaction by boiling the reaction mixture for 5 minutes (Hu et al, 2002).
The fluorescence is measured at two points: at excitation at 560nm and at emission at 590nm. 4-methylumbelliferyl phosphate (4-Mu-P) was used as the substrate of alkaline phosphatase-conjugated secondary antibody in the conventional ELISA. Here excitation was at 385 nm and emission at 448nm. Total tau levels are determined by the CSF sample before use in the assay. Dephosphorylation of the CSF sample was done before its use: “for 6 hours at 37°C in buffer, containing 50 mmol/L Tris- HCl, pH 8.0, 10 mmol/L MgCl2, 1% beta-mercaptoethanol and 0.5 mmol/L phenylmethylsulfonyl fluoride” (Hu et al, 2002). Total tau was calculated from the study with the recombinant human brain. Phosphorylated tau was calculated from ADP tau purified.
The ELISA with bienzyme substrate recycle was found 1300 times more sensitive in the detection range than the conventional ELISA (Hu et al, 2002).
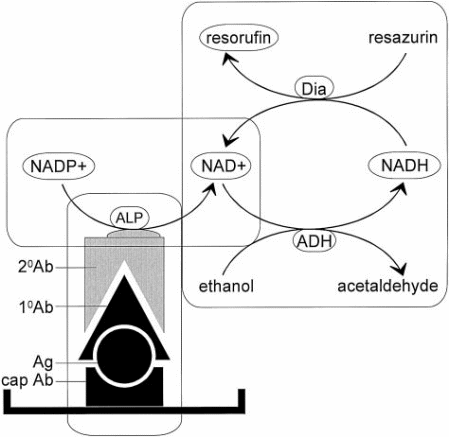
Diagram showing the principle of the bienzyme-substrate-recycle ELISA. Alkaline phosphatase, which is linked to secondary antibody, dephosphorylates NADP+ to NAD+. Then, NAD+ enters a highly NAD+ specific redox cycle, in which NAD+ is reduced to NADH by alcohol dehydrogenase, and the NADH produced is oxidized back to NAD+ by diaphorase with the concomitant reduction of resazurin (a nonfluorescent substrate) to resorufin (a fluorescent product). The resorufin accumulates with each cycle of NAD+ NADH- NAD+ and the fluorescence of resorufin is measured at 560 nm excitation and 590 nm emission. (Adapted from Hu et al, 2002)
Results
Buerger’s study showed that the CSF levels of P-tau 231P were related to the NFT count and HP tau load in all the neocortical brain regions but not the allocortical hippocampus (2006). There was an association between the neuritic plaques and the P-tau 231P in the frontal lobe. Increased disease duration was associated with enhanced NFT count, NP count, and HP tau load for the frontal cortex. CSF levels of P-tau 231P were related to the P-tau 231P of the brain homogenate of the frontal cortex.
The phosphorylated tau protein epitopes for threonine 181, serine 199, and threonine 231 have similar diagnostic results (Blennow and Hampel, 2003). These may be useful for future research. Fifteen percent of patients with mild cognitive impairment progress to AD but the follow-up of these MCI patients need to go on for a longer period of 5 years before sure information is obtained. This study decided that CSF markers are found insufficient concentrations in AD and can differentiate from mild cognitive impairment (Blennow and Hampel, 2003).
The ratio of phosphotau to Abeta42 was found to be significant in AD patients (Maddalena et al, 2003). This criterion was sufficient to differentiate AD cases from normal healthy subjects who were used as controls, dementias which were non AD and people with other neurological disorders (Maddalena et al, 2003).
Hampel too found that all p-tau subtypes were increased in patients with AD (2004). Correlations were not seen among the three types. Age had no relationship with the subtypes. AD could be differentiated from frontotemporal dementia by using p-tau 231 and p- tau 181.
Conclusion
This analytic review of the studies focused on the relationship of hyperphosphorylated tau proteins to the development of Alzheimer’s disease with specific details which would provide an idea of the pathophysiology of Alzheimer’s disease. Biochemical changes in the CSF affect the brain due to the proximity of the brain and extracellular space where the CSF is found (Blennow and Hampel, 2003). The biomarkers would reflect the pathogenesis occurring like neuronal degeneration, deposition of amyloid-beta peptide in the plaques and hyperphosphorylation of the tau proteins, and the formation of the tangles (Blennow and Hampel, 2003). The measurement of the combination of CSF phosphorylated tau protein and Abeta42 has been studied as biochemical markers (Maddalena et al, 2003). The amount of hyperphosphorylated tau proteins in CSF is maximum for severe degenerating diseases like Creutzfeldt-Jakob disease and less in AD (Otto et al, 1997 cited in Blennow and Hampel, 2003). The classification of illnesses into Alzheimer’s disease, frontotemporal dementia, and major depression may rely on the results of Hampel et al’s study (2004). Hampel’s study was the first to use three different assays on the same group of patients. The hallmark of AD is the neurofibrillary tangles believed to be due to the hyperphosphorylated tau proteins (Buerger, 2006).
Future research needs to establish CSF p-tau proteins as potential biomarkers for routine diagnosis of AD (Hampel et al, 2004). A study is already underway in this direction. Neuroimaging is starting and a set of different potential biomarkers is being studied in the National Institute of Aging Initiative on Neuroimaging in Alzheimer’s Disease: it is a five-year longitudinal study. The possible core marker would be the measurement of CSF p-tau proteins (Hampel, 2004). The ratio of phosphotau to Abeta42 could not be related to the severity of AD (Maddalena, 2003); future studies should be done using independent groups of patients. Ideally, the patients in the study should be followed till postmortem when a histopathological confirmation must be done (Maddalena et al, 2003). The study should not have selection biases as has occurred in Maddalena’s study. Studies are difficult in primary care settings as lumbar puncture for collection of CSF is not feasible. Future studies must focus on using the ratio of phosphotau to Abeta42 for monitoring disease progression (Maddalena et al, 2003). However, a limitation lies in performing lumbar punctures for so many participants. Atraumatic needles are available and the procedure has become smoother. Mere measurement of phosphotau and Abeta 42 may not be sufficient as a postmortem of AD brains show many lesions other than the tangles and the plaques. Studies in the future may show a biochemical marker pattern indicating a whole range of abnormal proteins deposited in the brain in AD (Maddalena et al, 2003). Studies have to evaluate whether a “combination of different p-tau epitopes or assays might improve diagnostic accuracy” (Blennow and Hampel, 2003).
References
Blennow, K. and Hampel, H. (2003). CSF markers for incipient Alzheimer’s disease. Lancet Neurology, Vol. 2, p. 605-613
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. (2006). Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin
sections and immunocytochemistry. Acta Neuropathol (Berl) 2006; [Epub ahead of print].
Buerger K, Hampel H. Evaluation of phosporylated tau protein as a core biomarker of Alzheimer’s disease. In: Iqbal I, Winblad B, editors.
Proceedings of the 9th International Conference on Alzheimer’s Disease and Related Disorders. Philadelphia, Pennsylvania: Alzheimer’s Association; 2004. p. 46–58.
Buerger, K., Ewers, M., Pirttila, T., Zinkowski, R. Alafuzoff, Teipel, S.J. et al, (2006) CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain (2006) 129, 3035–3041. Web.
Frank RA, Galasko D, Hampel H, Hardy J, De Leon M, Mehta DP, et al. (2003) Biological markers for therapeutic trials in Alzheimer’s disease. Proceedings of the biological markers working group; NIA initiative on neuroimaging in Alzheimer’s disease. National Institute on Aging Biological Markers Working Group. Neurobiol Aging 2003; 24: 521–36.
Hampel H, Buerger K, Zinkowski R, Teipel SJ, Goernitz A, Andreasen N, et al.(2004) Measurement of phosphorylated tau epitopes in the differential diagnosis of Alzheimer disease: a comparative cerebrospinal fluid study. Arch Gen Psychiatry 2004; 61: 95–102.
Hampel H, Bu¨rger K, Pruessner JC, Zinkowski R, DeBernardis JDK, et al.(2005). Correlation of cerebrospinal fluid levels of tau protein phosphorylated at threonine 231 with rates of hippocampal atrophy in Alzheimer disease. Arch Neurol 2005; 62: 770–3.
Hu,Y.Y., He, S.S., Wang, X., Duan, Q.H., Grundke-Iqbal, I. and Iqbal, K. et al, (2002). Levels of Nonphosphorylated and Phosphorylated Tau in Cerebrospinal Fluid of Alzheimer’s Disease Patients. American Journal of Pathology, Vol. 160, No. 4, April 2002 American Society for Investigative Pathology.
Maddalena, A., Papassotiropoulos, A., Mu ller-Tillmanns, B., Jung,H.H. Hegi, T. Nitsch, R.M.et al.(2003). Biochemical Diagnosis of Alzheimer Disease by Measuring the Cerebrospinal Fluid Ratio of Phosphorylated tau Protein to Beta-Amyloid Peptide42 Arch Neurol. 2003;60:1202-1206.
Wolf, H., Jelic, V., Gertz, H.J., Nordberg, A., Julian, P., Wahlund, L.O. (2003). A critical discussion of the role of neuroimaging in mild cognitive impairment., Acta Neurologica Scandinavia, Vol.107, Supplement 179, p. 52-76.