Introduction
It is noteworthy that there are at least five explanatory theories for the origin and spread of HIV. Among them is the idea that the virus originated from a flawed oral polio vaccination that was administered somewhere in Africa in the 1950s. there is also another theory that AIDS is, in fact, a weapon of mass destruction that was invented in a lab in a ploy of biological welfare but it soon got out of hand and the perpetrators lost control of this critical creation. Regardless of the numerous theories that have been created over the years to attempt to explain the occurrence of AIDS, what remains as a fact is the particular intelligence applied by the HIV once it sets upon the body.
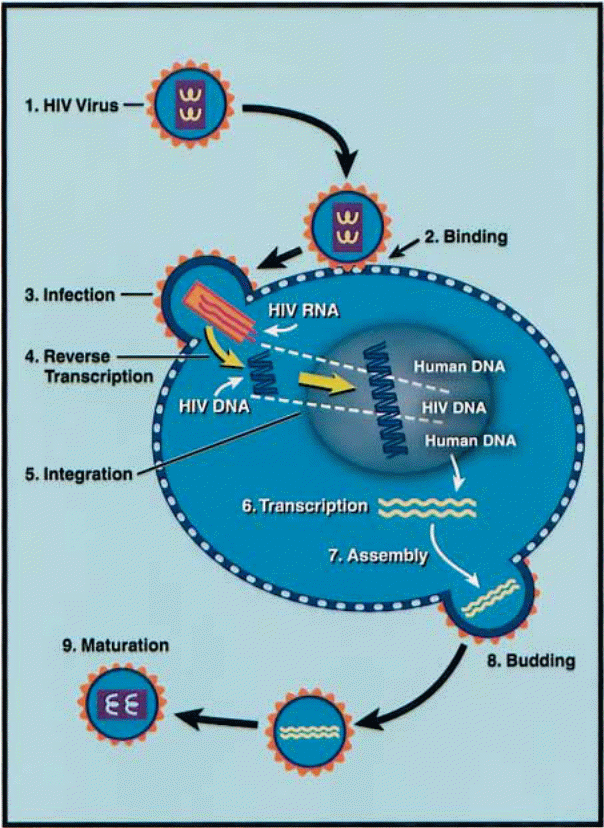
In effect, what becomes clear in the field of virology (research on viruses) is that first, the virus invades a cell and permeates the plasma membrane. Subsequently, with the use of the complementary DNA (cDNA) that has already been artificially synthesized by the viral RNA (mRNA) upon the intrusion of the host cell membrane through reverse transcription, the viral regulatory proteins Vif and Vpr assist in nucleic acid transportation. The cDNA is inscribed into the host DNA leading to the production of various viral products. Later, the pre-integration complex is exported to the host cytoplasm and again with the aid of the secreted viral proteins, as well as cytoplasmic communication channels, the HIV virion is populated or assembled and later churned out of the infected cell.
HIV is a genius or intelligent enemy to the human immune system. For a simple explanation of what occurs in the body, the researcher resorts to an allusion to human warfare to explain the attack and conquer tactics applied by HIV. The HIV attacks the body in the form of mRNA, which attacks the nucleus of the T cells and immediately replicates its DNA using the material in the host cells. Subsequently, the DNA produces a protein in the form of TAA, which is the virus that has been replicated. Thus, another T cell may be attacked. Therefore, when the virus enters the body, the body’s immune system reacts by producing T cells, specifically CD4+ cells to fight the invasion. On the membranes of these CD4+ cells, there are protein nodes that act like locks to the cells. The virus has glycoproteins (GP) that mediate entry into the cell membrane of the host cells and these are gp 41 and up 120. Subsequently, these glycoproteins attach to the relevant nodes on the cell membrane and induce the production of the enzyme responsible for reverse transcription as well as integration into the cell.
Once in the host cell, the mRNA now attacks the nucleus and replicates itself. It then proceeds to produce DNA and then a protein that is a virus. This leads to confusion of the white blood cells, which are inherently wired to fight antibodies and thus have the relevant intelligence that is required to aid that fighting because fellow white blood cells have now started producing the virus. The virus then attacks the white blood cells as it continues to reproduce. In the end, the CD4+ count reduces and a person is said to be HIV+. When the CD4+ count reaches 250, the person becomes an AIDS patient. It is interesting to note that HIV does not kill people with AIDS. Such persons are likely to die from secondary infections of other diseases such as tuberculosis, or cancer, or even a simple cough (Chinen, & Shearer 2002, p. 190).
What the virus does is that it weakens the immunity of such a person leading to the person’s susceptibility to successive illnesses and when they are infected with these illnesses, their immunity is too weak to battle the same. Antiretroviral Drugs or ARVs are thus engineered to battle the virus at two primary levels (Centres for Disease Control 1997, p. 165). The first is at the transcription stage. Therefore, the ARV would reduce the production of the enzyme responsible for reverse transcription, which means that the replication of HIV viruses in the body would be contained. At the second level, the ARV would work on the nucleus of the host cell (Moore, & Chaisson 1999, p. 1935).
However, there are latent shortcomings in these antiretroviral drugs because whereas they may reduce the replication of HIV, they do not address the virus that has already been released into the body. This has led to the conducting of various probability tests where hypothetically there would be HIV ARVs invented to deal with HIV reservoirs in the body. As of 1999, the study indicated that it would take at least 60 years or six decades of ARV therapy for the complete clearance of the virus from the host (Zhang, Ramratanam, Tenner-Racz, He, Vesanen, & Lewin et al. 1999, p. 1612).
Secondly, because HIV mutates randomly and a single mutation has been known to affect the entire system, making it immune to treatment, it means that a patient would have to use a large number of drugs to balance out the virus replication and personal cell functionality. The related side effects have been known to demoralize and/or de-motivate patients to the point of giving up antiretroviral therapy.
Epidemiology: Statistical Representation of HIV Infection Worldwide and Explanation of the Data
Overview of Global HIV Situation
The importance of acquainting ourselves with the worldwide statistics of HIV infection cannot be understated. This epidemiological perspective is especially useful in the facilitation of more efficient and effective prevention and treatment efforts. A simple illustration of how this works can be found in reviewing the infection pattern. Those people who are living with the virus, and are not aware of their positive status may refrain from unprotected sex if they were to know that they were infected. Moreover, they could start treatment early, thereby reducing the number of AIDS-related deaths annually. On the other hand, those that are not infected with HIV could obtain an education on responsible sexual behavior that is aimed at improving sexual behavior.
This would reduce the incidence of HIV infection. Moreover, pregnant women could also be targeted to ensure that they receive the safest delivery programs and services to protect their children from infection at birth ( UNAIDS 2011) and (World Health Organisation 2010). In short, awareness forms a great weapon in the ongoing fight against HIV infection. However, it is not possible to spread this awareness if the parties responsible for sensitizing the public are unaware of what they are dealing with. In other words, they are unaware of the situation on the ground and hence the reason why the epidemiology of AIDS and HIV infection becomes a critical area of study.
In 2007, there were 33.2 million people living with AIDS worldwide. The adults made up for 30.8 million of this round figure. The remaining 2.5 million were children under the age of 15. Also, 15.4 million of the adults were women living with AIDS. The number of newly infected in 2007 was 2.5 million and 2.1 million of these were adults with 400000 being children under the age of 15. Still, in 2007, there were 2.1 million deaths related to AIDS with 1.7 million being among the adults and 330000 being children under the age of 15. A critical analysis of these figures indicates an increase taken together of HIV-infected persons because, in the year 2007, more people were newly infected than the number of people that died due to AIDS-related causes. Subsequently, in 2011, the United Nations Political Declaration on HIV and AIDS was passed and the participants had in mind ten targets that they aspire to achieve with regards to curbing this epidemic by 2015 (UNAIDS 2011). Among the targets set out by the various UN-member states include:
- 50% reduction of HIV infection through sexual transmission.
- 50 % reduction of HIV infection among needle users that share needles when injecting drugs (Mathers et al. 2008, p. 1738).
- Eliminating the incidence of newly born children being infected by HIV and the number of mothers that die due to AIDS-related causes.
- Reaching a 15 million ceiling of the people that have access to ARV therapy.
- 50 % reduction of Tuberculosis (TB) deaths among people living with AIDS (Suthar et al. 2012, p. 270).
- Ensuring that low and middle-income countries arrive at the US $ 22 Billion to the US $ 24 Billion HIV AIDS resource investment ceiling.
- Elimination of gender-based inequity and disparity in the administration of HIV infected patients by ensuring that fundamental rights and freedoms are upheld in each member state.
- Ensuring the integration of HIV management into global health and development initiatives by eliminating parallel systems presently associated with HIV services that make the epidemic a unique entity and in most instances fuels stigmatization.
Upon the ratification of this declaration by UN member states in 2012, the subsequent year marked the first reporting period (UNAIDS 2012). Respective member states were given various reporting systems that were aimed at analyzing how the individual nation was doing especially in respect to HIV prevention and treatment. Of the 196 UN member states, 186 reported biennially in 2012. This made it apparent that global health initiatives can indeed receive unanimous support if handled with the appropriate amount of urgency and collaborative effort necessary. In 2012, 34 million people were living with AIDS (UNAIDS 2012). In 2012, the number of new infections declined radically by 2.5 million. This was 20 % lower than in 2001. In 2011, there were 1.7 million AIDS-related deaths worldwide, which was a 24 % decline from the 2.3 million that died in 2005 due to AIDS-related causes.
The cause for this notable decline in AIDS-related deaths can be attributed to increased awareness among the nations of the world regarding HIV and AIDS. This awareness spreads to an awareness of the availability of prevention and treatment options in the sense of condom use and abstinence as well as ARV therapy. This has gone a long way in increasing the life span of HIV infected and AIDS patients by more than 10 years. Additionally, it has reduced the number of new infections. Overall, the number of people that end up dying from AIDS is cut by more than 50 %. At the global level, various measures have been undertaken to curb the epidemic. A manifestation of these exerted efforts is in the political commitment expressed by various countries in the implementation of national policies that are in line with various UN declarations such as those that prohibit AIDS-related discrimination. Secondly, most states have put in place policies that are aimed at eradicating gender-based inequalities by upholding the fundamental freedoms and rights of respective members of the society.
Sub Saharan Africa
This region is made up of 69% of HIV-infected persons worldwide. In other words, one in every 20 people (or 4.9%) is HIV positive. The decline in the rate of new infections was second highest in the Caribbean and stood at 25 % (UNAIDS 2012). This translates to a compound figure of 1.8 million new infections in comparison to the year 2001. The deaths that occur due to AIDS-related causes in Sub-Saharan Africa have declined by 32 % in the period spanning 2005 to 2011. However, a point of concern, which becomes apparent when analyzing Sub Saharan Africa, is that the region is also the region that is responsible for 70 % of all AIDS-related deaths worldwide. It is also interesting to note that in the United States, where a large chunk of the population spots black Americans who are of African descent there is racial variability in susceptibility to infection.
African Americans form the majority of HIV infected individuals. Studies looking into the cause of this disparity in terms of racial profiling with HIV infection cannot find any biological indication for their susceptibility to the disease. Currently, the notion that is held by most researchers is that their sexual behaviors are more promiscuous due to perhaps sociological factors affiliated with the race (Brady, Friedman, Cooper, Flom, Bempalski, et al. 2008, p. 328).
A close review of these statistics makes it obvious that Africa is the region that is worst affected by the AIDS pandemic. However, Sub Saharan Africa is on its way to recovery albeit at a snail’s pace, as compared to North Africa, which seems to be retrogressing concerning HIV prevalence, and prevention and treatment efforts.
It has become apparent that by increasing efforts to promote ARV therapy administration, the number of incident cases also reduces. This means that there is an inversely proportional relationship between these two variables and that an increase in the number of patients receiving ART (Antiretroviral Therapy) results in a decrease in the number of people becoming newly infected with HIV. This was manifested in South Africa where HIV infection rates dropped by 17 % for every 10 % increase in those receiving ART (Johnson et al. 2012, p. 1552). What these figures indicate is that scaling up ART caused a reduction in new HIV infections (WHO 2003). However, these figures are not an indicator of the exact cause for this occurrence; neither can they be reversed as the number of new infections has nothing to do with the number of AIDS patients accessing ART. Recently, several awareness campaigns and treatment programs have been carried out in sub-Saharan Africa and notable changes in risky behaviors have been observed in most parts, mostly in Kenya, Malawi, Mozambique, Namibia, Zambia, and Nigeria.
The Middle East and North Africa
These two regions are exhibiting an interesting trend in statistical representation. Whereas among the rest of the world countries are reporting a marked decline in the number of new infections, which is proof of the success of efforts to curb new infections, in the Middle East and North African regions, the incidence of new infections is on the increase. In the 2012 reporting, the Middle East and North African nations recorded that the number of new infections had increased by 35 % (UNAIDS 2012). This reflected more than 37000 new infections from the 27000 infections recorded in 2011. The number of AIDS-related deaths increased by 17 % in 2011.
Caribbean, Eastern Europe, and Central Asia
Approximately, 1 % of adults in total are living with AIDS in these regions. This number has been constant for the past two decades (Downs, Heisterkamp, Rava, Houweling, Jager, & Hamers 2000, p. 2180). The Caribbean displayed the sharpest decline in the incidence of new infections by 42 % (AIDS Action, 2007). In Eastern Europe and Central Asia, the number f new infections began to escalate in the late 2000s after a marked period of stability spanning several years (Booth, Kwiatkowski, & Brewster 2006, p. 2219). In the Caribbean, 48% of the people living with AIDS passed on in 2011. In Asia, 4 % of the AIDS patients died due to AIDS-related causes, whereas in Eastern Europe and Central Asia, the rate of related mortalities increased by 21 %.
The United States
HIV was first diagnosed in the United States in the early 1980s. During this initial discovery period, the primary demographic composition of those infected was flawed and they were known to be white, gay, or bisexual men based in urban centers (Centres for Disease Control and Prevention 1982, p. 507). In 1983, more than three-quarters of 75 % of those that were diagnosed with HIV were men who had sex with other men, or MSM (UNAIDS 2005). This figure dropped dramatically as it became evident that even heterosexual partners could become infected with the virus (Centres for Disease Control and Prevention 1983). In 2007, only 47% of the new infections were male-to-male partners (Centres for Disease Control and Prevention 2009). In the Oceanic region, 41 % of those living with AIDS passed on due to AIDS-related causes. In Latin America, 10 % of AIDS patients died due to AIDS-related causes (Cohen 2006). In Eastern and Central Europe and North America, the number of AIDS-related deaths increased by 1 %.
It is noteworthy that the Americas presently stand on very solid grounds in terms of curbing the AIDS pandemic. Among the various reasons for this phenomenon is their rigorous health initiative that has been adopted in light of the UN Political Declaration on HIV and AIDS. This health initiative encompasses the various components that the UN Declaration on HIV & AIDS had in mind for the 2015 deadline (UNAIDS 2012). Among the recommended reform measures is behavioral change. This encompasses the various respective tasks or initiatives of ensuring condom availability; facilitating male circumcision, initiating programs aimed at educating sex workers and men having sex with other men (MSM) (Sanchez, et al, 2007, p. 581), as well as increasing the accessibility of ARV therapies to those infected with HIV (Allen et al. 2006, p. 99). Upon the implementation of these behavior change programs, it becomes apparent that they are successful upon the administration of various measurement techniques. Among the applicable measurement, regimes include the observance of the number of youths that get involved in early sexual conduct, the number of sexual partners that each sexually active person engages, and the rates of correct and consistent use of condoms among sexually active persons.
HIV Virology
Structure and organization
The Human Immunodeficiency Virus or HIV is a retrovirus with an affinity for CD4+ T Cells and monocytes. There are several types of white blood cells in the body, or T Cells and one such type is the CD4+. Infection is characterized by a decline in the T cell count and function, which subsequently results in a weak immune system. Additionally, B Cells become dysfunctional. This dysfunctionality is characterized by polyclonal activation. Next, the antibodies lose their specificity in terms of responses to various attacks and hypergammaglobulinemia is another likely occurrence.
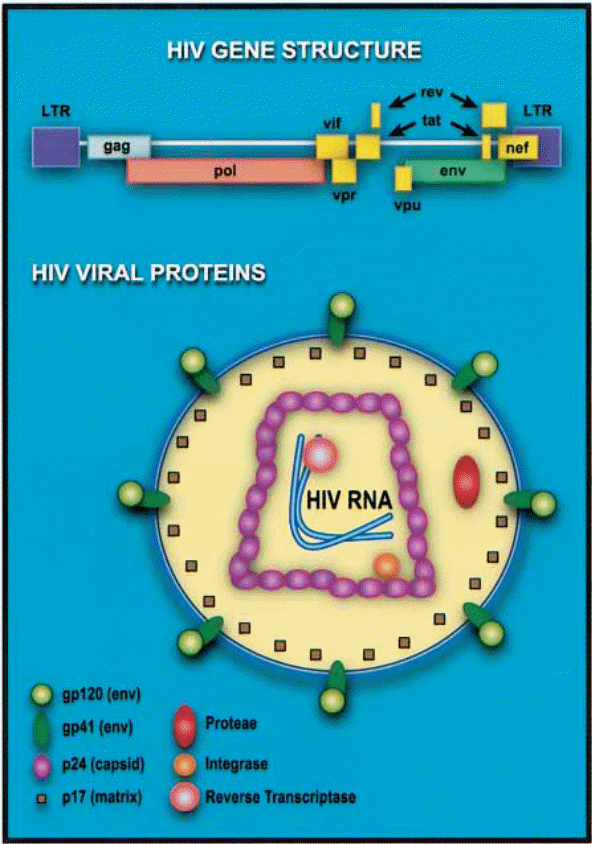
HIV is a lentivirus. The biological description is that HIV belongs to the family: Retroviridae; the sub-family: Lentivirinae; and the genus Lentivirus.
Since it belongs to the family of retroviruses, it is noteworthy that it shares the most notorious characteristic of retroviruses, which is that of containing independent ribonucleic acid (RNA) genome enclosed in a capsid and lipid envelope. The viral envelope comprises two layers. It is generated from the host cell during the assembly process just before the budding of the newly generated or coded virus occurs (Freed, & Martin 1995, p. 270). When the HIV enters into the host cell plasma, it comes with a viral pre-protein gp160. This glycoprotein later breaks down into other glycoproteins and viral regulatory proteins that work to facilitate the replication and transportation of HIV viruses in the host. However, at this initial stage, through a process of enzymatic cleavage, the gp 160 breaks down to form gp 41 and gp 120, which are the viral glycoproteins primarily charged with facilitating viral entry (into target cells) and syncytium formation. A syncytium is a mass of cytoplasm that contains numerous nuclei.
It is enclosed within a cell membrane. Syncytium formation by infected cells in the epithelium reticulum is one of the primary evidence of infection (Dorr, Westby, Dobbs, Griffin, Irvine, & Macartney et al. 2005, p. 4724). This occurs upon the clustering of infected cells and in the past, virology researchers have attributed this trait to the tropism of HIV type 1 virus to these cells. However, new research has indicated the very real possibility that this assembly of infected cells could be a mechanism employed by the virus to breed, grow, and develop infected cells as opposed to being a tropism.
Therefore, the classification of strains of HIV based on tropism is at best inconclusive and remains open to new developments. These developments have also shaped the type of therapies administered to patients with HIV (Fatkenheuer, Pozniak, Johnson, Plettenberg, Staszewski, Hoepelman, et al. 2005, p. 1172). However, there is unanimous agreement that the blockage of the virus from cell entry is the best stage to combat the virus considering that upon entry, it exhibits a rare intelligence whose complexity is exacerbated by the random mutations of the virus (Striski 2008, p. 99). Such matters make in-the- cell combating of the virus difficult and improbable as a final resort.
Of particular importance at this stage is the gp120, which contains a 5′ v3 loop that is responsible for the strong immune response by the viral cell.
Inside the HIV virus are three primary structural proteins, namely p24, p16, and p9. “The p24 protein forms the capsid that encloses the genomic RNA strands and the viral enzymes while the gp 16 is a matrix protein meaning that its primary function is the linkage of the viral core and the viral envelope membrane” (Levin, Mitra, Mascarenhas, & Musier-Forsyth 2010, p. 756). It is attached to the inner visage of the wrapper. It remodels nucleic acid structures to come up with the most thermodynamically stable stratifications (Levin et al. 2010, p. 754).
This image depicts the integrated DNA genome, which is also known as the provirus and is arrived at through DNA splicing. This provirus exists in this form and can thus remain for extensive lengths of time as the 5′ end triggers the replication of viral RNA in the normal course of host cell transcription. The newly transcribed RNA is either translated into the relevant viral proteins while still in the nucleus or it is transported out of the nucleus while still not spliced for packaging into newly produced virions.
It is also noteworthy that the “differences in viral structural architecture are directly related to the differences in the degree of virulence and transmissibility” (Chinen, & Shearer 2002, p. 190)(Point yet to be expounded further upon further research especially about other non-HIV viruses and their characteristic features). Additionally, these varying architectures are also responsible for the existence of various strains of HIV although, at this point, the respective identities of env and gag gene sequences play a critical role. The primary classification of HIV thus results in three strains, namely: M – Majority; O – Outliers; and N – Non – Majority-Non – Outliers. Under the majority strain, there are ten clades alphabetically labeled A up to J. “Subtype ‘B’ is most prevalent in the United States and Western Europe” (Chinen, & Shearer 2002, p. 190). Subtypes A, C, D, and E are common in the developing world. It is noteworthy that subtype E records relatively higher transmission levels among heterosexuals than B.
Reproduction Method of HIV -1 Virus
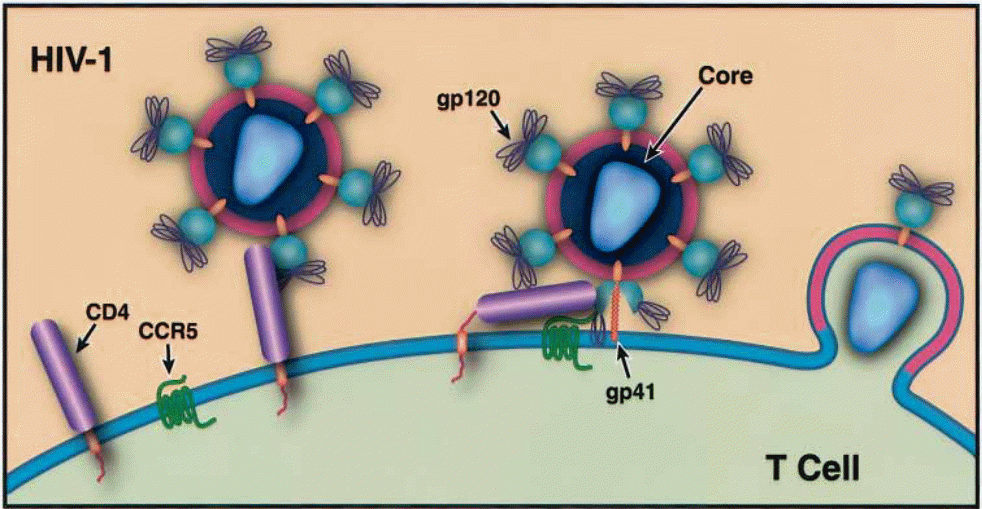
The HIV-1 virus enters the host cell by the use of glycoprotein 120 (gp 120) to initiate cognate recognition of certain proteins on the host cell surface (Messele, Brouwer, Girma, Fontanet, Miedema, Hamann, et al, 2001, p. 218). This gp 120 functions as a master key and unlocks the landing pad for the virus to attack and then embed itself in the host cell (Klatzmann, Champagne, Chamaret, Gruest, Guetard, Hercend, et al. 1984, p. 768). Since the host cell’s receptors of the viral load are specific proteins, the number of cells that can be infected by the virus is limited to those that have the requisite proteins on their cell surfaces. An example of such an entry point is the CD4 molecular surface because of the availability of the CD4 protein. This protein is amply held by cells on the T lymphocytes, monocytes, and macrophage lineages (Shehu-Xhilaga, & Oelrichs 2003, p. 10). The virus also manipulates the chemokine receptors, either CXR4 or CCR5 to gain access into other host cells (Dalgleish, Beverley, Clapham, Crawford, Greaves, Weiss, et al. 1984, p. 765).
Despite the original belief that the virus only attacks cells with CD4 expression, recent studies have indicated that even those cells without such expression can be and are invaded by the virus, with the assistance of chemokine receptors (Borsetti, Parolin, Ridolfi, Sernicola, Geraci, Ensoli, et al. 2000, p. 6689). Chemokine receptors are non – CD4 proteins that act as transmembrane co-receptors that are susceptible to HIV invasion (Liu, Soda, Shimisu, Haraguchi, Jinno, Takeuchi, et al. 2000, p. 279). There is a class of seven transmembrane receptors that can be formed by a wide range of proteins in the event of spatial proximity, which would also provide the doorway for HIV entry into a cell but the most common are CCR5 and CXCR4 (Saha, Zhang, Gupta, Dave, Yimen, & Zerhouni 2001, p. 67). This fact became apparent when it became apparent that some cells that had nonexistent CD4 expression were also being invaded. Therefore, it was clear that there were other conditions that made body cells susceptible to HIV invasion (Choe, Farzan, Sun, Sullivan, Rollins, Ponath, et al, 1996, p. 1144) and (Deng et al. 1996, p. 663). Under this classification of entry points, another classification of HIV becomes evident. This involves the principle of cell tropism.
Consequently, there are two strata and these include the macrophage-tropic (M- Tropic) and the T cell tropic (T Tropic) strains. The M tropic strain uses CCR5 as the coreceptor at the cell level. As such, its viruses are known as R5 viruses (Cheng-Mayer, Liu, Landau, & Stamatatos 1997, p. 1659). They target macrophages, primary T cells, and (minimally) CD4+ T Cell lineages. They are easier to transmit sexually than their T tropic counterparts (Yu, McLane, Ratner, O’Brien, Collman, Essex, et al. 1994, p. 10238). T tropic strain uses the CXCR4 as coreceptors at the cell membrane level, therefore they are commonly christened X4 viruses (Cho, Lee, Carney, Berson, Doms, & Martin 1998, p. 2511).
Their targets in the host are primarily the cells that express CD4+ (T cells). The viruses then work through encoding various viral proteins, which eventually induces the formation of syncytia in the invaded host. It is also noteworthy that in the initial stages of the infection, it’s the R5 viruses that are predominant in the host. However, in advanced stages, both R5 and X4 occur. Just like it is with the CD4 protein expression correlating with the host cell’s susceptibility to infection, so it is with the amount of chemokine receptor expression and susceptibility of the host cell to infection or invasion.
Moreover, recent strains of the virus (mutants) are exhibiting invasion tactics that do not require either CD4 or chemokine receptor expression so long as there is CD8 expression on the cell membrane (Dragic, Litwin, Allaway, Martin, Huang, Nagashima, et al. 1996, p. 671). To counter this new development is the proposition for the use of CD4 binding molecules to distort their appearance and thus prevent cognitive capacities of the virus that would induce entry. Another possibility is the use of beta-chemokines and peptides or T genes that resemble and/or block gp41 functioning upon encounter (Este, & Telenti, 2007, p. 85)and (Kilby et al. 1998, p. 1304). Also noteworthy is the fact that in the event of the mutation of CCR5, the cells become protected from HIV infection (Robertson 2003, p. 470). Such mutations usually do not affect the host negatively because soon enough, other chemokine receptors adopt the functions of CCR5. This is perhaps the second avenue that further studies are exploring as a possibility for retroviral therapy.
Upon entry, the viral gp 41 binds with the cell’s plasma membrane (heparan sulfate). This causes the viral envelope to fuse with the plasma membrane. Upon fusion, the capsid is released into the cytoplasm. Subsequently, “the viral capsid discharges its RNA and other elements into the host cytoplasm” (Chinen, & Shearer 2002, p. 190). With the assistance of the various proteins mentioned above as regulatory proteins, the process of reverse transcription of the viral RNA ensues and the end product is the complementary deoxyribonucleic acid cDNA. The conditions maintained for successful reverse transcription include the presence of a primer (t RNA molecule) which is sourced from the host cell’s cytoplasm. The enzymatic activity involved in reverse transcription causes a degradation of the viral RNA template. It is interesting to note that the “reverse transcriptase incorporates an incorrect nucleotide every 1500- 4000 bases” (Chinen, & Shearer 2002, p. 190).
This explains the numerous mutations of HIV and subsequent drug resistance of the virus. The enzyme reverse transcriptase facilitates transcription of deoxyribonucleic acid (DNA) from RNA, through a reverse process known as transcription. What makes this even more interesting is that in the case of HIV – 1, this entire process occurs in the lymphocyte, monocytes, or macrophages, which form part of the defense mechanism of the host. This, in turn, contributes to the confusion and dysfunctionality of the immune system because the new white blood cells are confused as fellow lymphocytes are now producing viruses and they upon infection begin to produce viruses. In the end, immunodeficiency occurs, making the infected individual highly susceptible to secondary infections. However, recent studies are looking into the adoption of non-nucleoside and nucleoside inhibitors of the enzyme reverse transcriptase on the premise that if it can be inhibited at this level, the virus can be prevented from replicating.
The next stage of invasion involves the transportation of the newly synthesized HIV – 1 cDNA to the nucleus with the assistance of the Vpr and Vif proteins. Specifically, as noted above, the Vpr protein assists in the import of the HIV – 1 pre-integration complex into the nucleus. Upon encountering the nucleus membrane, the Vpr protein and matrix protein gp16 issue out a nucleus localization signal that causes them to be allowed in. On the other hand, the Vif is effectual in the increment of the virus’s infectious potential by facilitation of its association with cytoskeletal elements. In the nucleus, the viral cDNA initiates abrupt assimilation with the main unit gene. The response is speeded up by the biological molecule namely integrase. At this point, it is also noteworthy that the developments of inhibitors for the enzyme integrase are underway. Immediately as the assimilation of the viral cDNA takes place into the harbor cell’s body, the natural processes that lead up to cellular dictation occur. However, since the DNA is infected, the outcome of cellular dictation now includes “low short levels of short, multiply spliced RNA transcripts” (Chinen, & Shearer 2002, p. 190).
Tat (trans-activating) Proteins
They are early RNA binding proteins that are primarily responsible for regulating the transcription of the viral gene. This makes them important facilitators of viral replication. A unique feature of tat proteins is their ability to be released and/or enter cells freely while remaining active even outside the viral cytoplasm. This unique feature makes it ideal for the proteins to regulate several viral-specific genes. Therefore, it follows that inhibition of protein production results in a remarkable suppression of HIV viral replication in the body. Tat proteins “trans – activate transcription by binding the 5′ end of the viral DNA sequence” (Watson, & Edwards 1999, p. 1522). This single simple function is responsible for multiplying the rate at which viral transcription occurs by more than a thousandfold.
Rev (Regulator of Expression of Virion) Protein
Rev is a protein that is encoded virally (through an HIV gene). It is also sequence-specific and RNA-binding (Pollard, & Malim 1998, p. 493). The proteinous elements thereof are critical to the transport of materials across the nuclear envelope in the form of export and import between the nucleus and the cytoplasm of the host cell. This is affected by the indigenous cell transportation routes, which feature the relevant signals that mediate cellular transport from the nucleus to the cytoplasm. Consequently, in case the Rev is absent, the host RNA spicing mechanism conducts splicing quickly to ensure the production of Rev, Tat, and Nef proteins. However, in case Rev is present, it facilitates the transportation out of the nucleus of non – spliced mRNA to encourage the production of structural proteins (gp 41 and 120) and the RNA genome. In effect, this ensures a positive feedback mechanism for HIV replication and attack of host cells that keeps the rate of infection perpetually high (Gallo, Wong-Staal, Montagnier, Haseltine, & Yoshida 1988, p. 504).
Nef (Negative Regulatory Factor) Protein
Nef features the largest number of functions that induce changes within the host cell. It forms part of the class of virulence factors because it is responsible for the manipulation of host cellular functions to pave way for viral infection, replication, and survival. Among its various capabilities include the advancement of clinical disease (up to AIDS level), cytoskeletal remodeling, and signal transduction. Especially during the initial stages of infection, Nef plays a key role in the inhibition of the expression of CD4+ cells. This means that the number of lymphocytes being activated to resist infections is minimized by the presence of this viral protein in the host. It does this by downregulating these CD4+ cells so that they do not respond to T Cell Receptor (TCR) stimuli, which under normal circumstances is the trigger for the abundant release of warrior lymphocytes (Ranjan, & Jameel 2005, p. 317). This mechanism thus reduces the chances of host resistance at the onset of infection and gives the virus an optimum environment for replication and infection within the host (Sinclair, Barbosa, & Feinberg 1997, p. 3647).
The elements of the HIV viral nucleus initially are changed to pr 55. The Pr 55 is the gag protein that is in charge of the assembly and budding stages of the HIV type 1 virus at the plasma membrane. It is a preprotein that occurs as a long spliced gag mRNA. Therefore, it can be cleaved upon mutation. When it exists as Gag-Pol, mutational cleaving of the same culminates in the “production of the viral enzymes integrase, protease, and reverse transcriptase” (Holm, Weclewicz, Hewson, & Suomalainen 2003, p. 4815). The functions catalyzed by integrase and reverse transcriptase have already been addressed and they are the integration of the viral cDNA into the host cell genome and the transcription of viral RNA into cDNA respectively. The function of the viral enzyme protease is the regulation and modulation of the requisite cleavage processes for the spliced viral RNA strands to form the respective viral pre proteins. Consequently, noticeable repression of HIV duplication is apparent upon the hanging-up of the manufacture of viral enzyme protease.
When the Gag-Pol and Pr 55 combine, it is usually after production from two reading frames. The reading frames regulate the production of structural versus regulatory viral enzymes. The first reading frame encodes Pr 555 or gag and the second one encodes the Gag-Pol. The first frame is more efficient than the second. This sheds light on the more relevant components for viral functionality.
Meanwhile, the intermediate mRNA, upon cleavage, produces glycoprotein (gp) 160. This gp 160 is transported to the host’s cell membrane. Upon virion formation, secondary cleavage of the gp 160 culminates in the formation of viral proteins Vpu, Vpr, and Vif.
Vif (Viral Infectivity Factor) Protein
This HIV 1-encoded protein neutralizes the host cell’s antiviral pathway that would otherwise efficiently neutralize HIV (Marin, Rose, Kozak, & Kabat 2003, p. 1401). Research indicates that this antiviral pathway can singly neutralize the virus and is located in human T cells and several leukemic non-permissive T cells (Poignard, Sabbe, Pichio, Wang, Gulisia, Katinger, et al. 1999, p. 432). The antiviral pathway is lined with cells that secrete APOBEC3G alias CEM-15, which is an enzyme responsible for conferring the antiviral phenotype on permissive cells, thus allowing them to combat HIV by mutating viral nucleic acids. If Vif were absent, the hypermutation effected by the APOBEC3G would render the virus dead upon arriving at the next host cell (Stanley, Ehrlich, Short, Yunkai, Xiao, Yu, & Xiong 2008, p. 8660). However, the HIV encoded Vif down-regulates this enzyme through a two-pronged approach. On the one hand, it binds it and thus prevents permissive cells from taking it up. Secondly, it facilitates the secretion of SLQ (Y/F) LA that degrades and eventually annihilates APOBEC3G production (Marin, Rose, Kozak, & Kabat 2003, p. 1403). Further research into this antiviral pathway is indicative of probable drug discovery.
Vpu (Viral Protein Unique) Protein
Against the backdrop of the evidence from peer-reviewed research on the functioning of HIV, which makes it apparent is that most of the HIV proteins have an absolute lack of enzymatic functioning. Therefore, they only work to weaken the host cells to pave way for the virus’s purposes. The Vpu protein’s functions reflect this mechanism (Bour, & Strebel 2003, p. 1032). The Vpu protein essentially regulates the release of the viral particle, the viral load, and the CD4+ receptor’s expression (Greenough, Brettler, Somasundaran, Panicali, & Sullivan 1997, p. 123).
The Vpu gene in the HIV encodes the VPU protein. It works to bring about the degradation of CD4+ in the endoplasmic reticulum. It is also facilitative of the process of releasing the virion from the plasma membrane of an infected host cell (Bour, Schubert, & Strebel 1995, p. 1518). Concerning CD4+ degradation, it is noteworthy that the Vpu binds with the protein tail of the CD4+, which exposes the vision to the spatial proximity of the sequential range of amino acids between 402 and 420 in the CD4+ cytoplasm. There is a correlation between the susceptibility of various host proteins to form bonds with virion Vpu and the rate of CD4 degradation. However, it is still unclear how the Vpu protein and the CD4+ proteins interact on a molecular level to culminate in the degradation of CD4+ cells since it is now apparent that binding alone does not cause degradation (Bour et al. 1995, p. 1510).
Vpr (Viral Protein R) Protein
This viral protein has essential characteristics that make it indispensable in virus replication. Originally, Vpr protein was classified as an accessory protein, meaning that virologists had classified it as being non-essential to HIV replication in vitro (Doranz, Rucker, Yi, Smyth, Samson, Peiper, et al. 1996, p. 1152). However, recent studies indicate the essence of Vpr protein in vivo (Bukrinsky, & Adzhubei 1999, p. 43). It is a critical facilitator of the nucleus import of the pre-integration combination of HIV -1. It also facilitates virus replication in non-dividing cells including macrophages (Gallay, Hope, Chin, Trono 1997, p. 9828).
In those host cells that multiply fast (proliferating), Vpr induces infected – cell arrest at the G2 /M stage of cell division (Poon, Grovit-Ferbas, Stewart, & Chen 1998, p. 268). It ensures that the rate of host cell regeneration is minimal and that it is eventually cut out completely (Muthumani, Choo, Zong, Madesh, Hwang, Premkumar, Thieu, Emmanuel, Kumar, Thompson, & Weiner 2006, p. 174). This action causes immune system dysfunction. The Vpr protein acts as a stimulant for virus transcription in the cytoplasm of the host cell, and the regulation of activation and apoptosis of infected cells (Bukrinsky, & Adzhubei 1999, p. 45). It is also noteworthy that the Vpr protein does not work alone but with the aid of cellular proteins. It specifically has an affinity for those proteins with the WxxF base of amino acids. Vpr alters host cell transcription to foster the replication of HIV (Cohen, Dehni, Sodroski, & Haseltine 1990, p. 3098).
The Pre-Integration Complex
Upon invasion into a host cell, the HIV information is contained in the RNA genome. Subsequently, through reverse transcription (aided by enzyme reverse transcriptase, which comes in the viral package or particle), the viral RNA is transformed into a single strand DNA and later into a double-stranded DNA (Fouchier, & Malim 1999, p. 287). This process of self-priming is complex and has been explained above. It occurs with the aid of various cellular and viral proteins including matrix and integrase. Subsequently, the double-stranded DNA is relocated to the nucleus as a complex. It is noteworthy that the virus does not automatically overpower the body upon invasion. The body using various mechanisms valiantly fights initially viral replication.
This explains the extended asymptomatic phase of HIV in most people. Among the key warriors against HIV infection is the human tripartite motif 5 alpha (TRIM5alpha) (Stremlau, Owens, Perron, Kiessling, Autissier, & Sodroski 2004, p. 849). This cell factor is renowned for identifying the virus, specifically picking on the capsid and destroying it prematurely before the invasion (Stremlau, et al 2006, p. 5518). This sort of attack occurs even before reverse transcription can occur. However, the occurrence of this TRIM5alpha cell factor does not automatically mean that such a cell would disintegrate the HIV virion’s capsid. There are different strains of the cell factor and in most cases; they react differently to the expression by the virion (van Manen, Rits, Beugeling, van Dort, Schuitemaker, & Kootstra 2008, p. 18).
Assembly and Budding Processes
The elements Vpu, Vpr, and Vif work in tandem with some other harbor proteins from the cell’s plasma membrane to make a viral wrapper that then surrounds the shell of the virus, which bears viral RNA components. This assembly process requires a lot of energy that is believed to be sourced from unidentified host cellular activities. At this stage, the viral polypeptides have been formed but they are yet immature because they lack some critical components such as the viral envelope and the viral proteins. With the aid of the viral enzyme protease, these immature polypeptides become populated into the fully functional HIV RNA transcripts that are capable of encoding the necessary viral components. The primary structural immature viral RNA polypeptide is a gag, which is very capable of forming the requisite viral proteins through transcription. Moreover, gag can independently hijack host cell proteins and pathways to make its way to the plasma membrane where it shall again use the aid of host cell proteins to complete its formation into a viral particle. The final stage is the budding stage, which features the activity of gag through p6 protein that is deposited at the gag polypeptide C terminus.
This p6 protein reacts with the host cell’s tumor suppressor [101 (TSG101)] gene to facilitate the production of newly assembled virions or virus particles (Bieniasz 2006, p. 57). Initially, the immature particles are clustered as immature particles, and then with the aid of the enzyme protease, they are developed into mature particles during budding. After this stage, the Vpu protein comes in handy in the release of the viruses after this late replication stage from the cell membrane. At the cell membrane, there is a cell factor known as tetherin that naturally inhibits the release of viral particles from the cell (Poon, et al. 1998, p. 268).
The Vpu protein alters its function to release the matured virus particles from the cells and thereby allow them to invade other cells in the host. Once released from the cell, these functional viral particles are free to attack other cells in the body and therein carry out the virus replication process. Eventually, with the lapse of time, the virus invades even the lymph nodes themselves, and other vital organs that are responsible for the provision of immunity from various infections, leaving the body truly defenseless. In advanced stages, HIV develops into AIDs. Eventually, the patient develops viremia, which is a condition indicating the presence of the virus in the bloodstream. The next segment of this paper shall deal with immunology and shall demonstrate how the body’s immune system responds to the virus.
Immunology of HIV
Overview of HIV Immunology
The immune system has two options in terms of reaction to HIV infection. On the first turn, the humoral immune function acts as the major extracellular immune response and the cellular immune response covers the intracellular defense mechanism. Evidence of HIV – 1 infection is primarily manifest with the reduction or decline of B cells, which are a class of lymphocytes of white blood cells that have on their cell surface BCR expression. BCR stands for B cell receptor and is a protein that allows the B cells to react in a customized manner to respective infections. There is a range of lymphocytes in the human anatomy and the rest include T cells and NK – Natural Killer cells. This segment of the thesis shall be dealing with how the immune system reacts to a viral invasion from the onset. It is noteworthy that chemokines are perhaps a critical variable in the development of an HIV vaccine because they have been noted to prevent vertical transmission from infected mothers to HIV exposed uninfected children.
What was interesting in this particular study was that the newly born babies did not indicate increased production of specific HIV cytotoxic cells but instead tended to secrete more chemokines (De Rossi 2002, p. 56). The exact inhibitor used in such cases is the Leukemia inhibitor factor, which inhibits HIV viral replication, and is thus injected into the placenta. Studies of HAART (Highly Active Antiretroviral Therapy) responsive HIV-positive patients and non-progressors (those patients that have been diagnosed with HIV infection but who exhibit minimal to nonexistent symptoms over their lifetimes -asymptomatic) indicate that certain conditions are indispensable if the virus is to be kept at bay. These include a plurality of TH1 type cytokines such as IL – 2 and IFN – a vigorous response by CD4+ T Cells, similarly vigorous responses by cytotoxic CD8+ T Cells, and an increase in the synthesis of Type A chemokines (Grabar, Le Moing, Goujard, Egger, Leport, Kazatchkine, et al. 2005, p. 289).
Upon invasion, the host body notably applies the above mechanisms to rebut HIV infection and replication. However, the degree of application of the said responses varies among individuals depending on several factors, including genetic makeup.
The HIV responds in like manner by:
- creating a variance on the immune sites. This prevents effective immune responses and thus allows the virus to replicate rapidly. It is also noteworthy that as the virus replicates, the internal viral processes include more than 10000 mutations within the life span of the virus. Therefore, they can easily confuse and overwhelm the immune system.
- Reduction of MHC on the cell surfaces and
- Reducing the number of CD8+ T Cells (Gao, Nelson, Karacki, Martin, Phair, Kaslow, et al. 2001, p. 1689).
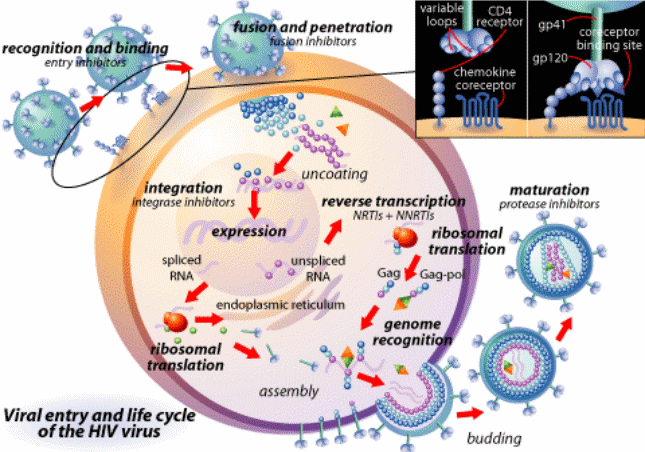
Humoral Immune Response to HIV Infection
HIV viruses do not replicate in B cells. However, they cause severe B cell dysfunction by viral protein mediating toxicity and cytokine dysregulation. The effects of such intrusion by the virus are made manifest in the host in the form of B Cell hyperplasia, circulating immune complexes, an increase in the number of autos –antibodies, and polyclonal hypergammaglobulinemia among other symptoms. Eventually, they culminate in the depletion of B cells, leaving the immune system vulnerable to opportunistic infections. At this point, the B cells can no longer produce the specific antibodies required by the body to combat HIV by the recall (memory) and new antigens as well as T- dependent and T – independent antigens (Gorse, Patel, Belshe 2001, p. 1175). The compound effect of these processes is the failure in the production of specific antibody responses, which correlates with an increase in the production of non-specific immunoglobulin G, A, and M.
Patients exhibiting high viremic levels also manifest a subpopulation of B cells that have a low CD 21 expression. This condition translates t the enhancement of secretion of immunoglobulin and malfunctioning antibody responses and could be part of the explanation for the collapse of the humoral immune system upon HIV infection.
More specifically, the HIV glycoprotein 120 (gp 120) tampers with the functioning of B cells by binding the VH3 domain of the membrane immunoglobulin in like manner to the way superantigens operate. When HIV encounters a mononuclear cell, it uses specific antibodies to launch an invasion. This in turn leads to an increased rate of phagocytosis of opsonized particles. When the HIV encounters matches, it can “activate them via the classical and alternative pathways that are inherently used by the host processes” (Holm, Weclewicz, Hewson, & Suomalainen 2003, p. 4815). The host usually responds to viral invasion by depositing complement C3on the viral membrane. However, complements C5 through C9 are poor at membrane level attack. The host secretes soluble C D 16, which is an inhibitor of the C3 receptor-mediated HIV 1 infection of monocytes. To inhibit or better yet, block viral fusion, virologists have suggested the administration of passive IgG3 that is more potent than IgG1 in vitro (Schwarf, Golding, King, Eller, Frazier, Golding, et al. 2001, p. 6561).
However, this strategy is not without risk because they (virologists) have also deduced that such administration of passive antibodies singles out viral replication processes and the virus reacts by mutating, leading to viral escape mutants that are drug-resistant (Borrow, P, Lewicki, H, Wei, X, Horwitz, Pffeifer, & Meyers 1997, p. 207).
Cellular Immune Response
HIV Infection induces a powerful cellular immune response to curb the infection. However, this does not address the problem fully because, whereas the virus may be combated at the cell level, the replication process goes uninhibited. Therefore, the matter of HIV reservoirs is not addressed (Fahey, Taylor, Detels, Hofmann, Melmed, Nishanian, Giorgi, et al. 1990, p. 1886). The virus thus evades the immune system by various evasive maneuvers. These ways include “The latency of the HIV provirus, sequestration reservoirs, later evolution of the viral strain in advanced stages as it progresses from R5 to X4, the down regulating of MHC molecules, the epitope mutations of various viral proteins, and the favorable regulation of the Fas ligand” (Rowland-Jones, Dong, Fowke, Kimari, Krause, & Newell 1998, p. 1759)
As noted above, it is possible to gauge and measure the progressive state of the disease by counting the total of CD4 + T Cells as well as CD8 + T Cells (Margolick, Jonnenberg, Chu, O’Gorman, Giorgi, & Munoz 1998, p. 266). The CD4 + T cells are also known as helper cells. This is because they come to the aid of CD8 + T cells in the establishment of memory. They also “produce cytokines as well as interact with other immune cells to facilitate the wholesome immune response” (Koup, Safrit, Cao, Andrews, McCleod, Borkowsky, et al. 1994, p. 4654). Specific cytotoxic CD8 + T cells kill the viruses in the body upon enlisting the assistance of CD4+ T cells, which prime the CD8+ T cell responses and maintain an immunologic memory as well as cytolytic or killer response.
As CD8 + T cells are depleted, viremia becomes apparent in HIV infected patients. With the onslaught of an acute infection, the levels of CD8 + T cells in the body can increase 20-fold in reaction to the invasion. Subsequently, the disease usually depletes in record time. The CD8 + T cells in an HIV infected area fight back infection by secreting a soluble protein, which is culpable for inhibiting HIV replication. Consequently, even before the antibodies associated with HIV infection become apparent in an infected patient, it is possible to detect the specific cytotoxic CD8 + T cells in the body. Nevertheless, as the disease progresses, their numbers get depleted, although at no one time shall they ever be negative or totally absent in the body, including even when peripheral CD4 + T cells are completely depleted. In an acute infection, they form 8% of the immune defense and in chronic infections, they manifest at the rate of 1-2 % (Spiegel, Ogg, DeFalcon, Sheehy, Monard, Haslett, et al. 2000, p. 1019).
It is also noteworthy that the reduction of the sum total of T cells is not an instantaneous process following infection but more of a gradual decline and annihilation. Initially, they develop abnormalities such as lymphoproliferative responses to antigens and mitogens. Studies conducted among nonprogressors and those patients that respond positively to HIV treatment indicate a trend of positive lymphoproliferative response to gag proteins exhibited by a decrease in the viral load as well as the fortification of TH1 cytokine production (Gea-Banacloche, Migueles, Martino, Shupert, McNeil, et al. 2000, p. 1088). Another trend of HIV is the preference to infect memory CD4 T cells (CD45RO+). This becomes even more interesting when it is apparent that despite this preference, it is the naïve cells (CD45RA+ and CD62L+) that get depleted faster (Bonnet, Thiebaut, Chene, Neau, Pellegrin, Mercie, et al. 2005, p. 202). A possible explanation for this discombobulating phenomenon could be the chronic stimulation of memory cells, which possibly leads to their latency.
Upon the initiation of ART, the rate of opportunistic infections takes a nosedive. Some virologists have attributed this to a restricted T cell repertoire. However, this is not necessarily accurate because incident infections, as well as lymphomas, become common in such patients. This means that longer durations of treatment therapies are necessary in order to restore the T cell repertoire. The HIV virus has various means of depleting T cells and these include induction of apoptosis (programmed cell death); impairment of production; and redistribution of the T cells (both infected and uninfected) into the lymph glands and organs where there are other viruses.
The apoptosis could be through several means including env mediation; Vpr induced cell G2 arrest; disrupting the cell membrane syncytia formation; enlisting the aid of the natural killer and cytotoxic cells’ cytolysis processes; and auto-immune reactions. According to Franco, Leon-Leal, Leal, Cano-Rodriguez, Pineda, and Macias, et al. (2000, p. 496), “The X4 strain of HIV 1 infection infects the thymus and in so doing destroys the stromal support as well as distorting the secretion of cytokine. On the bone marrow level, several effects of HIV infection can be felt, including anemia, neutropenia, and thrombocytopenia”. However, these bone marrow dysfunctionalities all get reversed upon the initializing of ART. It is also noteworthy that dendritic cells in the body can attach the HIV and transport it into the lymph gland or organs without getting infected (Lisziewicz, Gabrilovich, Varga, Xu, Greenberg, Arya, et al. 2001, p. 7627).
Innate Immunity
In the late 1980s, it became apparent that HIV infected patients with a certain CD8+ T lymphocyte phenotype could secrete a protein known as CAF. The presence of this protein in their bodies made them highly resistant to the effects of HIV infection. In later years, virologists have used gene chip technology and antibody tagging techniques in experiments through which they have found out that HIV infected patients have the expression of three genes: alpha – Defensin 1, -2, and -3, which were stimulated upon infection (Chattergoon, Boyer, & Weimar 1997, p. 761).
Subsequently, it has been discovered that upon synthetic preparation of these proteins, the rate of HIV replication is significantly inhibited in vitro. Over a long time, it was perplexing to most virologists that many sex workers who contracted the virus seemed to be more resistant to its effects than other patients who were not equally promiscuous. This even led to the development of a myth that they were immune to HIV. However, further studies have indicated that the cause of this resistance is attributable to high CD8+ T Cell counts as opposed to their capacity to produce HIV specific antibodies. Consequently, most of the research is now revolving around factors that stimulate CD8+ T cell production as a possible source of vaccination.
Human Leukocyte Antigen (HLA)
It became obvious to virologists that there was a correlation between the presence of HLA alleles and the rate of disease (AIDS) progression in HIV infected patients. Subsequent studies made it apparent that a minimal change in the composition of an HLA molecule in the form of an amino acid change can either positively or negatively affect the progression and development of AIDS and can affect the efficiency of the ART (Hendel, Caillat-Zucman, Lebuanec, Carrington, O’Brien, et al. 1999, p. 6945). The HLA B35 has been identified as the most prevalent allele in HIV infection progression into the development of AIDS. On the other hand, alleles like B51, B27, and A2 have resonated with ART and their presence usually guarantees optimum clinical results in the administration of ART (Kassutto & Rosenberg 2004, p. 1451). Nevertheless, it is noteworthy that the absence or presence of the respective HLA phenotypes does not have any bearing on the patient’s susceptibility to contract HIV.
Cytokines
As noted above, HIV- 1 graduate with time from the R5 strain onto the X4 strain (Verrier, Burda, Belshe, Duliege, Excler, Klein, et al. 2000, p. 10029). This change is also reflected in the cytokine orientation in the host. As AIDS progresses, there is s switch from TH1 to TH2. TH1 is responsible for the induction of cellular immune response. This works to minimize the onslaught of viremia in the host (Brigido, Rodrigues, Casseb, Custodio, Fonseca, Sanchez, et al. 2004, p. 193). Studies indicate that upon infection, HIV – 1 induces the secretion of IL – 1, IL – 2, TNF – α, IFN – y, and IL 12 among others at various levels of the AIDS progression (Honda, Kitamura, Misutani, Oishi, Arai, Okura, et al. 1990, p. 4060). The exact manner of the effect of cytokines on disease progression is still ambiguous. However, a notable effect of the addition of cytokines in vitro is the restoration of natural killer (NK) cells. Perhaps it could be said that cytokines play a part in the existence of killer cells in the body. The IL – 7 favors AIDS persistence in the host by inhibiting the apoptosis of thymocytes and lymphocytes so that they now act as reservoirs for the viruses.
Chemokines
The chemokine receptor CCR5 is more prevalent (8-fold) in TH1 cells than in TH 2 cells. Conversely, the chemokine receptor CXCR4 is more prevalent (4 – fold) in T H 2 cells than in TH1 cells. However, another interesting phenomenon is expressed in the replication rates of the respective viruses. R5 viruses replicate better in TH2 cells. This possibly explains the tropism of the HIV phenotype in infected patients of progression from R5 to X4 as HIV advances to AIDS. As noted above, the CD8 + T cells are primarily responsible for the secretion of β chemokines, including RANTES and (Mitogen Lymphocyte Proliferation) MIP – 1 β as well as MIP – 1 α. These exist as natural ligands in CCR5 and they inhibit viral replication (Cocchi, DeVico, Garzino-Demo, Ayra, Gallo, & Lusso 1996, p. 1812). On the other hand, CXCR4 only stimulates SDF – 1 α as the natural ligand. This serves to block HIV entry by inhibiting host cell interaction with glycoprotein 120 (Oberlin, Amara, Bachelerie, Bessia, Virelisier, Arenzana, Seisdedos, et al. 1996, p. 834).
Naturally, chemokine therapy is one of the areas that are being pursued as a probable solution for inhibiting viral entry. Nevertheless, it is also apparent that large concentrations have to be applied for the effectiveness of treatment. This correlates with increased side effects as well as the likelihood of drug resistance.
HIV Treatment and Vaccine Development
Drugs
Most antiretroviral drugs work by inhibiting viral replication. This gives the patient a fighting chance at recovery. However, they do not address the HIV reservoirs that have been created by the virus and that continue to attack uninfected cells. Additionally, due to the numerous mutations of HIV, drug resistance is a real threat in HIV infected patients. To combat the problem of drug resistance, HIV pharmacologists are now focusing on the non – variable aspects of the virus that are conserved to avoid mutation prospects. Additionally, they are administering a variety of similar drugs in small amounts. The problem comes when the most effective drug therapy would require large concentrations, such as in the case of chemokines that inhibit the entry of HIV into the cell. Presently, HIV 1-infected patients are undergoing HAART, which stands for Highly Active Antiretroviral Therapy.
This therapy is characterized by the above drug resistance management suggestions. The HAART is a cocktail of drugs that inhibit RTase (Moore, & Chaisson 1999, p. 1941). Several options of Antiretroviral Therapies are under investigation, and these include nucleoside and non-nucleoside inhibitors (Gulick, Ribaudo, Shikuma Lustgarten, Squires, Meyer, et al. 2004, p. 1859). Nucleoside inhibitors act as an inhibition to RTase and therefore, bind the active site on the enzyme. This in turn brings to a stop the synthesis of 5′ and 3′ (Lythgo 2004, p. 85). On the other hand, non-nucleotide inhibitors bind the enzyme without the active sites causing it to become useless. The effects of these inhibitors are greatly significant in vitro. However, they are too specific. Therefore, any slight mutation of the virus usually renders them ineffective. Consequently, they are a therapy that is administered alongside other drugs (Weller, & Williams 2001, p. 1411).
Since drugs address viral aspects that would have the most debilitating effect on the virus, another likely target is the viral enzyme protease, which is responsible for the cleavage of glycoprotein 160. This enzyme is essential for attaining viral maturity (Blumenzweig, Baraz, Friedler, Danielson, Gilon, Steinitz, & Kotler 2002, p. 834). By blocking the protease substrate site, this cleavage cannot get underway. Another angle of drug therapy could take the mediation of viral entry into host cells, which glycoprotein 41 forms an attractive prospect. A synthetic factor called T – 20 that is prevalent in hydrophobic alpha-helices can easily bind the critical ends of the gp41 domain thus inhibiting binding or fusion with the host cell (Kilby, Hopkins, Venetta, DiMassimo, Cloud, Lee, Alldredge, Hunter, Lambert, Bolognesi, Matthews, Johnson, Nowak, Shaw, & Saag 1998, p. 1304). This prevents viral entry. Other drug options include the inhibition of viral protein integrase using integracycin (Singh, Zink, Heimback, Genilloud, Teran, Silverman, Lingham, Felock, & Hazuda 2002, p. 1125) to prevent the transfer of viral complementary deoxyribonucleic acid to the host genome, and the deactivation of accessory proteins such as Rev by translucent genes (Mautino, Keiser, Morgan, 2001, p. 3593).
Vaccine Development
The objective of HIV vaccine development is the reduction of the spread of HIV as well as the absolute elimination of the virus’s presence in hosts’ bodies. The ideal solution would be that of immunostimulatory vaccines because these would enable the host body to combat the virus independently (Mascola, & Nabel 2001, p. 493). Additionally, such a vaccination option would be cost-friendly, easy to administer, and to store. Countless vaccines have been tested but very few have attained worthiness status to justify clinical trials. Most of the rest are not safe and they fail at the critical indicator of worthiness because they cannot elicit strong-enough CD4+ and CD8+ T cell responses. Subsequently, only one-third of the vaccines are permitted to clinical trial stages.
The various mechanisms of vaccine development include:
The use of Antigens
Several probable antigens can be used in vaccine development including env, glycoproteins 120, and 160, 120 meters, and V3 peptides. Secondly, vectors could be used in the delivery of antigens and these include alphaviruses, polioviruses, and herpes viruses among others.
The development process includes the use of a plasmid in the encoding of env and Rev genes. Studies have indicated that these respective genes stimulate a strong T cell response as well as secrete many chemokines that inhibit HIV viral replication.
Combined Administration
As vaccine development evolves, later stages involve the joint administration of antigen encoding plasmids other cytokines as well as immunostimulatory sequences of the deoxyribonucleic acid.
Conclusion
This thesis has looked into the epidemiology, virology, and immunology of HIV infection, as well as the drug and vaccine development options available for the same. From the study conducted, it is apparent that although it may yet take a while, it is indeed possible that the epidemic that is AIDS can be managed and controlled. The United Nations came up with a strategy to be implemented by the respective member states to ensure 50 % eradication of most of the HIV variables by 2015. This includes sexual transmission, transmission via injections among drug users sharing needles, antidiscrimination and integration policies, as well as awareness campaigns and treatment and vaccine options were addressed by the 2011 Political Declaration on HIV and AIDS.
Since the declaration was made, 186 of the 196 United Nations members reported via the mechanisms and technologies employed by the UN and the World Health Organisation (WHO) on the status of HIV in their respective nations. This has unified global data on the pandemic. Out of the various nations that reported the data on the global occurrence of HIV to indicate that it is possible to present a united front in the fight against HIV / AIDS and that more effective results shall be derived through such methods. One person cannot achieve issues such as the financial objective of raising the US $ 24 Billion as an AIDS resource and that no nation can safely barricade its borders to infected persons worldwide. Therefore, unity is critical for policy success.
Secondly, on virology, HIV displays a unique intelligence in its infection or invasion strategy. It is a very complex virus because it has the viral complementary DNA that it then uses to replicate itself upon entry. It also has individual parts to carry out the various respective functions necessary for HIV persistence and progression to AIDS. Consequently, finding an individual solution that would solve all basic steps of infection and replication is near impossible, hence the lack of a cure. The situation is further aggrieved by the occurrence of more than 10000 mutations in the life span of each virus. However, presently, there are several highly effective ARTs in place that include the use of nucleoside and non-nucleoside inhibitors, inhibitors for the respective viral enzymes and accessory proteins, and the development of immunostimulatory DNA. Immunological responses to the virus are either humoral or cellular. At the failure of these, HIV progresses fast to AIDs and ultimately to death but before that, viremia can be used to judge progression.
References
AIDS Action 2007, State facts: HIV/AIDS in Puerto Rico, Word Press, Washington, DC.
Allen, C, et al. 2006, ‘Sexually transmitted infection use and risk factors for HIV infection among female sex workers in Georgetown’, Journal of Acquired Immune Deficiency Syndromes, vol. 43 no. 1, pp. 96–101.
Bieniasz, P 2006, ‘Late budding domains and host proteins in enveloped virus release’, Virology, vol. 344 no. 1, pp. 55-63.
Blumenzweig, I, Baraz, L, Friedler, A, Danielson, U, Gilon, C, Steinitz, M & Kotler, M 2002, ‘HIV-1 Vif-Derived Peptide Inhibits Drug-Resistant HIV Proteases’, Biochemistry and Biophysics Research Communication, vol. 292 no. 2, pp. 832-840.
Bonnet, F, Thiebaut, R, Chene, G, Neau, D, Pellegrin, J, Mercie, P, et al. 2005, ‘Determinants of clinical progression in antiretroviral-naive HIV-infected patients starting highly active antiretroviral therapy. Aquitaine Cohort, France, 1996-2002’, HIV Medicine, vol. 6 no. 3, pp. 198-205.
Booth, R, Kwiatkowski, F & Brewster, J 2006, ‘Predictors of HIV sero-status among drug injectors at three Ukraine sites’, AIDS , vol. 20 no. 17, pp. 2217–2223.
Borrow, P, Lewicki, H, Wei, X, Horwitz, S, Pffeifer, N, & Meyers, L1997, ‘Antiviral pressure exerted by HIV1 specific cytotoxic T-lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus’, Nature Medicine , vol. 3 no. 1, pp. 205-11.
Borsetti, A, Parolin, C, Ridolfi, B, Sernicola, L, Geraci, A, Ensoli, B et al. 2000, ‘CD4-independent infection of two CD4(-)/CCR5(-)/CXCR4(+) pre-T-cell lines by human and simian immunodeficiency viruses’, Journal of Virology, vol. 74 no. 14, pp. 6689-94.
Bour, S & Strebel, K 2003, ‘The HIV-1 Vpu protein: a multifunctional enhancer of viral particle release’, Microbes and Infection, vol. 5 no. 11, pp. 1029-39.
Bour, S, Schubert, U & Strebel, K 1995, ‘The human immunodeficiency virus type 1 Vpu protein specifically binds to the cytoplasmic domain of CD4: implications for the mechanism of degradation’, Journal of Virology, vol. 69 no. 3, pp. 1510–1520.
Brady, J, Friedman, R, Cooper, H, Flom, L, Bempalski, B, et al. 2008, ‘Estimating the prevalence of injection drug users in the U.S. and in the Large U.S. metropolitan areas from 1992 to 2002’, Journal of Urban Health: Bulletin of the New York Academy of Medicine, vol. 85 no. 3, pp. 323–351.
Brigido, L, Rodrigues, R, Casseb, J, Custodio, R, Fonseca, L, & Sanchez, M, et al. 2004, ‘CD4+ Tcell recovery and clinical outcome in HIV-1-infected patients exposed to multiple antiretroviral regimens: partial control of viremia is associated with favorable outcome’, AIDS Patient Care STDS., vol. 18 no. 4, pp. 189-98.
Bukrinsky, M, & Adzhubei, A 1999, ‘Viral protein R of HIV-1’, Reviews in Medical Virology, vol. 9 no. 1, pp. 39-49.
Centres for Disease Control 1997, ‘Update on trends in AIDS incidence deaths and prevalence—United States, 1996’, MMWR, vol. 46 no. 1, p. 165.
Centres for Disease Control and Prevention 1982, ‘Current Trends Update on Acquired Immune Deficiency Syndrome (AIDS) — United States’, MMWR, vol. 31 no. 37, pp. 507–508.
Centres for Disease Control and Prevention 1983, Acquired Immunodeficiency Syndrome (AIDS). Weekly Surveillance Report – United States. AIDS Activity, Centre For Infectious Diseases, Centres For Disease Control. Web.
Centres for Disease Control and Prevention 2009, HIV/AIDS Surveillance Report, 2007. Vol. 19. Atlanta: U.S. Department of Health and Human Services, Centres for Disease Control and Prevention. Web.
Chattergoon, M, Boyer, J, & Weimar, D 1997, ‘Genetic immunisation: a new era of vaccines and immuno therapeutics’, FASEB Journal, vol. 11 no. 1, pp. 753-63.
Cheng-Mayer, C, Liu, R, Landau, N, & Stamatatos, L 1997, ‘Macrophage tropism of human immunodeficiency virus type 1 and utilisation of the CC-CKR5 coreceptor’, Journal of Virolology, vol. 71 no. 2, pp. 1657-61.
Chinen, J & Shearer, W 2002, ‘Molecular virology and immunology of HIV infection’, Journal of Allergy and Clinical Immunology, vol. 110 no. 2, pp. 189 -198.
Cho, M, Lee, M, Carney, M, Berson, J, Doms, R & Martin, M 1998, ‘Identification of determinants on a dualtropic human immunodeficiency virus type 1 envelope glycoprotein that confer usage of CXCR4’, Journal of Virology, vol. 72 no. 3, pp. 2509-15.
Choe, H, Farzan, M, Sun, Y, Sullivan, N, Rollins, B, Ponath, P, et al. 1996, ‘The beta-chemokine receptors CCR3 and CCR5 facilitation of infection by primary HIV-1 isolates’, Cell, vol. 85 no. 7, pp. 1135-48.
Cocchi, F, DeVico, A, Garzino-Demo, A, Ayra, S, Gallo, R, & Lusso, P 1996, ‘Identification of RANTES, MIP-1alpha, MIP-1beta as the major HIV-1 suppressive factors produced by CD8 T-cells’, Science Journal, vol. 270 no. 1, pp. 1811-4.
Cohen, E, Dehni, G, Sodroski, J, & Haseltine, W 1990, ‘Human immunodeficiency virus vpr product is a virion-associated regulatory protein’, Journal of Virology, vol. 64 no. 6, pp. 3097-9.
Cohen, J 2006, ‘Up in smoke: epidemic changes course’, Science, vol. 313 no. 1, pp. 487–488.
Dalgleish, A, Beverley, P, Clapham, D, Crawford, D, Greaves, M, & Weiss, R, et al. 1984, ‘The CD4 (T4) antigen as an essential component of the receptor for the AIDS retrovirus’, Nature, vol. 312 no. 2, pp. 763-7.
De Rossi, A 2002, ‘Primary HIV infection in infants: impact of highly active antiretroviral therapy on the natural course’, Journal of Biological Regular Homeostatic Agents, vol. 16 no. 1, pp. 53-7.
Deng, H, Liu, R, Ellmeier, W, Choe, S, Unutmaz, D, & Burkhart, M, et al. 1996, ‘Identification of a major coreceptor for primary isolates of HIV-1’, Nature , vol. 381 no. 6584, pp. 661-6.
Doranz, B, Rucker, J, Yi, Y, Smyth, R, Samson, M, & Peiper, S, et al. 1996, ‘A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors’, Cell, vol. 85 no. 7, pp. 1149-58.
Dorr, P, Westby, M, Dobbs, S, Griffin, P, Irvine, B & Macartney, M et al. 2005, ‘Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity’, Antimicrboiological Agents of Chemotherapy, vol. 49 no. 11, pp. 4721-32.
Downs, A, Heisterkamp, H, Rava, L, Houweling, H, Jager J, & Hamers, F 2000, ‘Back-calculation by birth cohort, incorporating age- specific disease progression, pre-AIDS mortality and change in European AIDS case definition. European Union Concerted Action on Multinational AIDS Scenarios’, AIDS, vol. 14 no. 14, pp. 2179-89.
Dragic, T, Litwin, V, Allaway, G, Martin, S, Huang, Y, Nagashima, K, et al. 1996, ‘HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5’, Nature, vol. 381 no. 6584, pp. 667-73.
Este, J & Telenti, A 2007, ‘HIV Entry Inhibitors’, Lancet , vol. 370 no. 9581, pp. 81-8.
Fahey, J, Taylor, J, Detels, R, Hofmann, B, Melmed, R, Nishanian, P, & Giorgi, J, et al. 1990, ‘The prognostic value of cellular and serologic markers in infection with human immunodeficiency virus type 1’, New English Journal of Medicine, vol. 322 no. 26, p. 1886.
Fatkenheuer, G, Pozniak, A, Johnson, M, Plettenberg, A, Staszewski, S, & Hoepelman, A et al. 2005, ‘Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1’, Nature Medicine , vol. 11 no. 11, pp. 1170-2.
Fouchier, R, & Malim, M, 1999, ‘Nuclear import of human immunodeficiency virus type-1 preintegration complexes’, Advanced Virus Research, vol. 52 no. 1, pp. 275-99.
Franco, J, Leon-Leal, J, Leal, M, Cano-Rodriguez, A, Pineda, J, Macias, J, et al. 2000, ‘CD4+ and CD8+ T lymphocyte regeneration after anti-retroviral therapy in HIV-1-infected children and adult patients’, Clinical Exp Immunology, vol. 119 no. 3, pp. 493-8.
Freed, E & Martin, M 1995, ‘The role of human immunodeficiency virus type 1 envelope glycoproteins in virus infection’, Journal of Biology and Chemistry, vol. 270 no. 1, pp. 23883-6.
Gallay, P, Hope, T, Chin, D & Trono, D 1997, ‘HIV-1 infection of nondividing cells through the recognition of integrase by the importin/karyopherin pathway’, Procter National Academy Science USA , vol. 94 no. 18, pp. 9825-30.
Gallo, R, Wong-Staal, F, Montagnier, L, Haseltine, W, & Yoshida, M 1988, ‘HIV/ HTLV gene nomenclature’, Scientific Correspondence: Nature, vol. 33 no. 3, pp. 504.
Gao, X, Nelson, G, Karacki, P, Martin, M, Phair, J & Kaslow, R, et al. 2001, ‘Effect of a single amino acid change in MHC Class I molecules in the rate of progression to AIDS’, New English Journal of Medicine, vol. 344 no. 1, pp.1688-75.
Gea-Banacloche, J, Migueles, S, Martino, L, Shupert, W & McNeil, A, et al. 2000, ‘Maintenance of large numbers of virus-specific specific CD8 T-cells in HIV-infected progressors and non-progressors’, Journal of Immunology, vol. 165 no. 1, pp. 1082-92.
Gorse, G, Patel, G & Belshe, R 2001, ‘HIV type 1 vaccine-induced T-cells memory and cytotoxic T lymphocyte responses in HIV type-1 uninfected volunteers’, AIDS Research Human Retroviruses, vol. 17 no. 1, pp. 1175-89.
Grabar, S, Le Moing, V, Goujard, C, Egger, M, Leport, C & Kazatchkine, M, et al. 2005, ‘Response to highly active antiretroviral therapy at 6 months and long-term disease progression in HIV-1 infection’, Journal of Acquired Immune Deficiency Syndrome, vol. 39 no. 3, pp. 284-92.
Greenough, T, Brettler, D, Somasundaran, M, Panicali, D & Sullivan, J. 1997, ‘Human immunodeficiency virus type-1 specific cytotoxic T lymphocytes (CTL) viral load, and CD4 T-cell loss: evidence supporting a protective role for CTL in vivo’, Journal of Infection and Disease, vol. 176 no. 1, pp. 118-25.
Gulick, M, Ribaudo, J, Shikuma M, Lustgarten, S, Squires, K, & Meyer, W, et al. 2004, ‘Triple-nucleoside regimens versus efavirenz-containing regimens for the initial treatment of HIV-1 infection’, New English Journal of Medicine, vol. 29/350 no. 18, pp. 1850-61.
Hendel, H, Caillat-Zucman, S, Lebuanec, H, Carrington, M, O’Brien, S, et al. 1999, ‘New Class I and Class II HLA alleles strongly assoassociate with opposite patterns of progression to AIDS’, Journal of Immunology, vol. 162 no. 1, pp. 6942-6.
Holm, K, Weclewicz, K, Hewson, R, & Suomalainen, M 2003, ‘Human immunodeficiency virus type 1 assembly and lipid rafts: Pr55(gag) associates with membrane domains that are largely resistant to Brij98 but sensitive to Triton X-100’, Journal of Virology , vol. 77 no. 8, pp. 4805-17.
Honda, M, Kitamura, K, Misutani, Y, Oishi, M, Arai, T & Okura, T, et al. 1990, ‘Quantitative analysis of serum IL-6 and its correlation with increased levels of serum IL-2R in HIV-induced diseases’, Journal of Immunology, vol. 145 no. 1, pp. 4059-64.
Johnson, L, et al. 2012, ‘The effect of changes in condom usage and antiretroviral treatment coverage on human immunodeficiency virus incidence in South Africa: a model-based analysis’, Journal of the Royal Society Interface, vol. 9 no. 1, pp. 1544–1554.
Kassutto S & Rosenberg, E 2004, ‘Primary HIV type 1 infection’, Clinical Infection Discourse , vol. 38 no. 10, pp. 1447-53.
Kilby, J, Hopkins, S, Venetta, T, DiMassimo, B, Cloud, G, Lee, Y, Alldredge, L, Hunter, E, Lambert, D, Bolognesi, D, Matthews, T, Johnson, R, Nowak, M, Shaw, G, & Saag, M. 1998, ‘Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41- mediated virus entry’, Nature Medicine, vol. 4 no. 11, pp. 1302-1307.
Kilby, M, Hopkins, S, Venetta, M, DiMassimo, B, Cloud, G & Lee, J, et al. 1998, ‘Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry’, Nature Medicine, vol. 4 no. 11, pp. 1302-7.
Klatzmann, D, Champagne, E, Chamaret, S, Gruest, J, Guetard, D & Hercend, T, et al. 1984, ‘T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV’, Nature, vol. 312 no. 1, pp. 767-8.
Koup, R, Safrit, J, Cao, Y, Andrews, C, McCleod, G & Borkowsky, W, et al. 1994, ‘Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 infection syndrome’, Journal of Virology, vol. 68 no. 1, pp. 4650-5.
Levin, G, Mitra, M, Mascarenhas, A, & Musier-Forsyth, K 2010, ‘Role of HIV-1 nucleocapsid protein in HIV-1 reverse transcription’, RNA Biology, vol. 7 no. 6, pp. 754-74.
Lisziewicz, J, Gabrilovich, D, Varga, G, Xu, J, Greenberg, P, & Arya, S, et al. 2001, ‘Induction of potent human immunodeficiency virus type 1-specific T-cell-restricted immunity by genetically modified dendritic cells’, Journal of Virology, vol. 75 no. 1, pp. 7621-8.
Liu, H, Soda, Y, Shimisu, N, Haraguchi, Y, Jinno, A & Takeuchi, Y, et al. 2000, ‘CD4-dependent and CD4-independent utilisation of coreceptors by human immunodeficiency viruses type 2 and simian immunodeficiency viruses’, Virology, vol. 278 no. 1, pp. 276-88.
Lythgo, P 2004, Molecular Virology of HIV-1 and Current Antiviral Strategies. Web.
Margolick, J, Donnenberg, A, Chu, C, O’Gorman, M, Giorgi, J, & Munoz, A 1998, ‘Decline in total T-cell lymphocyte count: implications for AIDS pathogenesis’, Clinical Immunology and Immunopathology, vol. 88 no. 1, pp. 256-63.
Marin, M, Rose, K, Kozak, S, & Kabat, D 2003, ‘HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation’, Nature Medicine Journal, vol. 9 no. 11, pp. 1398-403.
Mascola, R & Nabel, G, 2001, ‘Vaccines for prevention of HIV disease’, Current Opinion on Immunology, vol. 13 no. 1, pp. 489-95.
Mathers, B, et al. 2008, ‘Global epidemiology of injecting drug use and HIV among people who inject drugs: a systematic review’, Lancet, vol. 372 no. 1, pp. 1733–1745.
Mautino, R, Keiser, N, & Morgan, R 2001, ‘Inhibition of Human Immunodeficiency Virus Type 1 (HIV-1) Replication by HIV-1 Based Lentivirus Vectors Expressing Transdominant Rev’, Journal of Virology, vol 75 no. 8, pp. 3590-3599.
Messele, T, Brouwer, M, Girma, M, Fontanet, L, Miedema, F & Hamann, D, et al. 2001, ‘Plasma levels of viro-immunological markers in HIV-infected and non-infected Ethiopians: correlation with cell surface activation markers’, Clinical Immunology, vol. 98 no. 2, pp. 212-9.
Moore, R & Chaisson, E 1999, ‘Natural history of HIV infection in the era of combination antiretroviral therapy’, AIDS, vol. 13 no. 1, pp. 1933-42.
Muthumani, K, Choo, A, Zong, W, Madesh, M, Hwang, D, Premkumar, A, Thieu, K, Emmanuel, J, Kumar, S, Thompson, C & Weiner, D. 2006, ‘The HIV-1 Vpr and glucocorticoid receptor complex is a gain-of-function interaction that prevents the nuclear localisation of PARP-1’, Nature Cell Biology, vol. 8 no. 2, pp. 170-9.
Oberlin, E, Amara, A, Bachelerie, F, Bessia, C, Virelisier, J, Arenzana, & Seisdedos, F, et al. 1996, ‘The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell line adapted HIV-1’, Nature, vol. 382 no. 1, pp. 833-5.
Poignard, P, Sabbe, R, Pichio, G, Wang, M, Gulisia, R, & Katinger, H, et al. 1999, ‘Neutralising antibodies have limited effects on the control of established HIV-1 infection in vivo’, Immunity, vol. 10 no. 1, pp. 431-8.
Pollard, V & Malim, M 1998, ‘The HIV-1 Rev Protein’, Annual Review of Microbiology, vol. 52 no. 1, pp. 491-532.
Poon, B, Grovit-Ferbas, K, Stewart, S, & Chen, I 1998, ‘Cell cycle arrest by Vpr in HIV-1 virions and insensitivity to antiretroviral agents’, Science, vol. 281 no. 5374, pp. 266-9.
Ranjan, S & Jameel, S 2005, ‘Biology of the HIV Nef protein’, Indian Journal of Medical Research, vol. 1 no. 1, pp 315-332.
Robertson, D 2003, ‘US FDA approves new class of HIV therapeutics’, Nature Biotechnology, vol. 21 no. 5, pp. 470-1.
Rowland-Jones, L, Dong, T, Fowke, R, Kimari, J, Krause, P & Newell, H 1998, ‘Cytotoxic T cell responses to multiple conserved HIV epitopes in HIV resistant prostitutes’, Journal of Clinical Investigation, vol. 102 no. 1, pp. 1758-65.
Saha, K, Zhang, J, Gupta, A, Dave, R, Yimen, M & Zerhouni, B 2001, ‘Isolation of primary HIV-1 that target CD8+ T lymphocytes using CD8 as a receptor’, Nature Medicine, vol. 7 vol. 1, pp. 65-72.
Sanchez, J, et al. 2007, ‘HIV-1, sexually transmitted infections, and sexual behaviour trends among men who have sex with men in Lima, Peru’, Journal of Acquired Immune Deficiency Syndrome, vol. 44 no. 5, pp. 578–585.
Schwarf, O, Golding, H, King, L, Eller, N, Frazier, D & Golding, B, et al. 2001, ‘Immunoglobulin G3 from polyclonal human immunodeficiency virus (HIV) immune globulin is more potent than other subclasses in neuneutralising HIV type 1’, Journal of Virology, vol. 75 no. 1, pp. 6558-65.
Shehu-Xhilaga, M & Oelrichs, R 2003, ‘HIV Management in Australasia a guide for clinical care’, Basic Virology, Immunology, and Natural History, vol. 1 no. 1, pp. 9-17.
Sinclair, E, Barbosa, P & Feinberg, M 1997, ‘The nef gene products of both simian and human immunodeficiency viruses enhance virus infectivity and are functionally interchangeable’, Journal of Virology, vol. 71 no. 1, pp. 3641-51.
Singh, B, Zink, L, Heimback, B, Genilloud, O, Teran, A, Silverman, K, Lingham, R, Felock, P & Hazuda, D 2002, ‘Structure, Stereochemistry, and Biological Activity of Integramycin, a Novel Hexacyclic Natural Product Produced by Actinoplanes sp. That Inhibits HIV-1 Integrase’, Organisational Letter, vol. 4 no. 7, pp. 1123-1126.
Spiegel, H, Ogg, G, DeFalcon, E, Sheehy, M, Monard, S & Haslett, P et al. 2000, ‘Human immunodeficiency virus type1- and cytomegalovirus-specific cytotoxic T-lymphocytes can persist at high frequency for prolonged periods in the absence of circulating peripheral CD4+ T cells’, Journal of Virology 2000 , vol. 74 no. 1, pp. 1018-22.
Stanley, B, Ehrlich, E, Short, L, Yunkai, Y, Xiao, Z, Yu, X & Xiong, Y 2008, ‘Structural Insight into the Human Immunodeficiency Virus Vif SOCS Box and Its Role in Human E3 Ubiquitin Ligase Assembly’, Journal of Virology 2008, vol. 82 no. 17, pp. 8656–8663.
Stremlau, M, Owens, C, Perron, M, Kiessling, M, Autissier, P & Sodroski, J 2004, ‘The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys’, Nature, vol. 427 no. 6977, pp. 848-53.
Stremlau, M, Perron, M, Lee, M, Li, Y, Song, B, & Javanbakht, H, et al. 2006, ‘Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor’, Procteur National Academy of Sciience USA , vol. 103 no. 14, pp. 5514-9.
Striski, J 2008, ‘Targeting HIV attachment and entry for therapy’, Advanced Pharmacology, vol. 56 no. 1, pp. 93-120.
Suthar, A et al. 2012, ‘Antiretroviral therapy for prevention of tuberculosis in adults with HIV: a systematic review and meta-analysis’, PLoS Medicine, vol. 9 no. 1, p. e1001270.
UNAIDS 2005, Evidence for HIV decline in Zimbabwe: a comprehensive review of the epidemiological data, UNAIDS, Geneva.
UNAIDS 2011, Countdown to zero: Global Plan towards the elimination of new HIV infections among children by 2015 and keeping their mothers alive. Web.
UNAIDS 2011, Political Declaration on HIV and AIDS: Intensifying Our Efforts to Eliminate HIV and AIDS Geneva. Web.
UNAIDS 2012, Global AIDS Response Progress Reporting 2012: guidelines – construction of core indicators for monitoring the 2011 Political Declaration on HIV/AIDS. Web.
UNAIDS 2012, Report on the Global AIDS Epidemic, UNAIDS, Geneva.
van Manen, D, Rits, M, Beugeling, C, van Dort, K, Schuitemaker, H & Kootstra, N 2008, ‘The Effect of Trim5 Polymorphisms on the Clinical Course of HIV-1 Infection’, PLoS Pathogens, vol. 4 no. 2, p. e18.
Verrier, F, Burda, S, Belshe, R, Duliege, A, Excler, J & Klein, M, et al. 2000, ‘A human immunodeficiency virus prime-boost immunisation regimen in humans induces antibodies that show interclade cross-reactivity and neutralise several X4- R5-and dual-tropic clade B and C primary isolates’, Journal Of Virology, vol. 74 no. 1, pp. 10025-33.
Watson, K & Edwards, R 1999, ‘HIV-1-trans-activating (Tat) protein: both a target and a tool in therapeutic approaches’, Journal of Biochemistry and Pharmacology, vol. 15/58 no. 10, pp. 1521-8.
Weller, I & Williams, G 2001, ‘ABC of AIDS: Antiretoviral Drugs. BMJ’, BioMedical Journal, vol. 322 no. 2, pp. 1410-1412.
WHO 2003, Scaling Up Antiretroviral Therapy In Resource Limited Settings:Treatment Guidelines For a Public Health Approach, World Health Organisation, Geneva.
World Health Organisation 2010, Antiretroviral therapy of HIV infection in infants and children: towards unviersal access.Recommendations for a public health approach: 2010 revision Geneva. Web.
Yu, X, McLane, M, Ratner, L, O’Brien, W, Collman, R & Essex, M, et al. 1994, ‘Killing of primary CD4+ T cells by non-syncytium-inducing Killing of primary CD4+ T cells by non-syncytium-inducing’, Proc Natl Acad Sci USA, vol. 91 no. 21, pp. 10237-41.
Zhang, L, Ramratanam, B, Tenner-Racz, K, He, Y, Vesanen, M & Lewin, S. et al. 1999, ‘Quantifying residual HIV-1 replication in patients receiving combination antiretroviral therapy’, New English Journal of Medicine, vol. 340 no. 1, pp. 1605-13.