Introduction
Sickle cell anemia is not as new as scientific documentation indicates. It was first documented in Africa in the 1870s and resulted in many deaths prior to the research that made the management of it more effective. While there is no cure for the disease, there are interventions and treatments available that help in minimizing and alleviating the symptoms of the disease. The following paper provides an overview of sickle cell anemia and current research on it to broaden the general understanding of this disease.
History
Hemoglobinopathy is the study of genetic abnormalities that involves the globin chains of hemoglobin, the red component in the blood that carries oxygen. The most common of these include the sickle cell trait. The first documented case of sickle cell disease was that of dental student Walter Clement Noel of Chicago in 1904, but symptoms associated with the disease are mentioned in African writings, referring to the condition as chwecheechwe, abututuo, nuidudui, and nwiiwii. When Noel was admitted, an intern named Ernest Irons noticed that Noel’s blood presented unusual features, that of many elongated cells. The attending physician, James Herrick, was informed, and they proceeded to document the progression of the disease over two-and-a-half years. It took Noel nine years to succumb, presumably as a consequence of his condition.
A researcher named Hall first coined the term “sickle cell,” but it was V.E. Emmel who documented that the sickle cell trait could be present in healthy individuals without developing anemia.
By the 1940s, it was established that the sickle cell was a result of abnormal hemoglobin but not the mechanism that led to the abnormality. In the 1950s, American scientist Vernon Ingram isolated the culprit, an amino acid called valine, which took the place of glutamic in the beta-globin chain of hemoglobin. It may sound like a trifling change, but it has had a profound impact on those who have it. (“Genetic basis of hemoglobinopathies- the history of the sickle cell”)
Causes
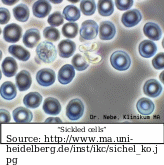
Sickle cell is a genetic disorder that turns red blood cells, which should be round and flexible, into stiff and sickle-shaped cells, which is why it is called a sickle cell. This is due to mutations in the beta-hemoglobin (HBB) gene.
Because they are inflexible, it is unable to travel smoothly in the blood vessels, bunching up and slowing the flow, preventing the efficient delivery of oxygen. This results in pain, organ damage, and infection. The anemia results from the early death of these cells with a life span of 20 days as opposed to the normal 120 days for normal cells. The body is unable to keep up with the supply of new cells (“BabyCenter editorial staff”).
It is transmitted via autosomal recessive inheritance, meaning it is passed from generation to generation and may present without any symptoms unless both parents carry the genes. In order for sickle cell trait to manifest into sickle cell anemia, a person must have two defective genes, one from each parent. In the US, it is commonly found in people of African, Spanish, Mediterranean, Middle Eastern, and Indian descent.
Diagnosis and Complications/Symptoms
The diagnosis of sickle cell anemia involves a blood test that looks for the presence of hemoglobin S, the form that underlies sickle cell anemia. Newborns are routinely tested for it, but adults and older children can also be tested. A positive screening will indicate the need for further testing that will determine if one or two genes are present. The sudden death of Missouri University (MU) football player Aaron O’Neal due to lymphocytic meningitis later indicated the involvement of sickle cell anemia in the condition. As a result, MU athletes, three of whom are already positive for sickle cell but are so far asymptomatic, are voluntarily subjecting themselves to screening procedures for the disease. This is based on the known effects of strenuous activity for people who have sickle cell anemia. A report from the National Collegiate Athletic Association claims that at least 15 athletes have died due to complications associated with sickle cell anemia. Those found to be positive for the sickle cell trait are asked to sign a medical waiver.
Sickle cell anemia presents with a variety of symptoms, including:
- Pain
- Enlargement of the spleen
- Damage to liver and kidneys
- Viral and bacterial infections as a result of a malfunctioning spleen
- Stroke from blocked blood vessels
- Aplastic crisis, which is when red cell production stops for ten days as a result of an infection
- Acute chest syndrome similar to pneumonia, when sickled cells get trapped in the lungs, eventually damaging the lungs
- Eye problems in the retina due to lack of oxygen, which may lead to blindness
- Jaundice
- Gallstones
- Open sores on the legs due to skin cells dying from lack of oxygen
- Painful erections or priapism for men due to sickled cells getting lodged in the penis, leading eventually to impotence
Treatment and medications
The only real potentially effective cure for sickle cell anemia is a bone marrow transplant, but this is not an option for the majority of the affected population because of the dearth of a suitable donor. Much of the treatment protocol focuses on preventing crises from occurring as well as relieving the effects of the symptoms and complications. The treatment protocols used for the disease include:
- Antibiotics, mostly penicillin for children from 2 months to 5 years of age
- Pain medications such as acetaminophen or stronger medication as well as anti-inflammatory medications.
- Hydroxyurea, a prescription drug normally used for cancer treatment, is believed to stimulate fetal hemoglobin that prevents the production of sickle cells.
- Blood transfusions using only red blood cells to increase the number of red blood cells in the system to alleviate the anemia to lessen the risk of stroke
- Supplemental oxygen through a breathing mask to alleviate acute chest syndrome
- Bone marrow transplant
- Surgery to correct vision problems or to remove a damaged spleen
There are some complications attached to some of these treatments. Regular blood transfusions tended to build up iron in the body which could also damage the heart, liver, and kidneys, so accompanying blood transfusions were regular detoxification of iron levels, usually using deferasirox, taken orally to eliminate excess iron administered to people more than two years old.
Bone marrow transplants are risky procedures because the protocol requires the use of chemotherapy to destroy the bone marrow, and then healthy marrow is inserted. There is a danger of rejection of the new marrow and could result in serious consequences to the patient. It also involves a long hospital stay as well as a prolonged round of anti-rejection drugs.
Recently, a lifelong sufferer of sickle cell anemia, 13-year-old Aaron Washington, underwent an experimental bone marrow transplant from donated bone marrow from her sister Tayla at Children’s Healthcare of Atlanta. While recovering, she suffered a stroke and seizure that led to the discovery of a hidden colon problem that had the potential for infection, a deadly occurrence for a recovering transplant patient. Doctors repaired the problem in the colon, and the stroke appeared to have caused minimal damage to Aaron. The early discovery of the colon problem was fortunate; however, Aaron is far from complete recovery.
Babies with both genes for sickle cell anemia are given an additional pneumococcal vaccine, PPV23, aside from the normal vaccines given to all babies.
Some treatments are experimental, such as gene replacement therapy, in which the defective gene is repaired and reinserted into the bone marrow. Another gene-based method is through deactivating the defective gene and reactivating another gene that is normally responsible for producing fetal hemoglobin. The inclusion of butyric acid, a food supplement that may encourage the production of fetal hemoglobin, is also being investigated. Another substance being considered for efficacy is clotrimazole, treatment for fungal infections, prevents the loss of water from red blood cells and will hopefully restrict the number of sickled cells that will form. Nitric oxide, which is low in a person suffering from sickle cell anemia, is supplemented by treatment with nitric oxide gas.
Prevention
Because the disease is transferred from generation to generation, the only real way to prevent it is to ensure that those carrying the recessive traits avoid conception if both parents are positive for the sickle cell trait.
In vitro fertilization, wherein the zygote is tested for the sickle cell gene before implantation, is an option for couples who want to have children but who are both at-risk for transmitting the disease to their progeny. However, the success of the procedure is not assured, and it can carry a hefty price tag.
Current statistics
In a study of over 700 patients over two decades conducted by University of Texas Southwestern Medical Center researchers Dr. Charles Quinn and Dr. Zora Rogers, the data showed that the chances of children with severe sickle cell anemia to reach the age of 18 are now at about 85%, a 12% increase from that of 30 years ago, due mainly to a more thorough understanding of the disease and advances in treatment protocols. Early screening of newborns also contributes to survival rates because treatment protocols are begun early before extensive organ damage makes it more complicated to treat. About 70,000 Americans suffer from the disease, and 50 years ago, half of them would not live beyond the age of 21. Of the 711 participants in the study, 15 had died from sickle cell anemia complications(“Sickle cell sufferers living longer, dying less from their disease”).
The risk of death can be predicted as well, researchers from Boston University School of Medicine (BUSM) and Boston University School of Public Health (BUSPH) claim using a network model they developed using five-year or more data from 3,380 adult and pediatric patients. This is significant for determining the appropriate treatment plan as well as providing statistical data for studying the pathophysiology of the disease.
Conclusion
Sickle cell anemia is a prolonged and painful lifetime condition that still has no viable cure even after extensive research on the disease. It is considered the most extensively studied genetic disease in medical history, and yet the process for reversing the mechanism that produces the abnormal hemoglobin has yet to be discovered.
Because it affects the blood, very little escapes being affected by the disease. Even the skin is affected if the disease is not properly managed. However, it is possible for sickle cell anemia sufferers to live a full life provided regular treatment and self-care protocols are religiously observed.
Works Cited
- “Genetic basis of hemoglobinopathies- the history of sickle cell.” Sickle Cell Anemia Foundation. 2007. Sickle Cell Anemia Foundation of America.
- “Sickle cell sufferers living longer, dying less from their disease.” The University of Texas Southwestern Medical Center at Dallas. 2004.
- BabyCenter editorial staff. “Sickle cell anemia.” BabyCenter.com. 2007. BabyCenter LLC. Web.
- Lange, Alex. “Sickle cell testing for athletes increases.” Columbia Missourian. 2007.
- Markiewicz, David. “Recovery difficult, but new treatment gives teen new hope.” ajc.com. 2007. The Atlanta Journal-Constitution. Web.
- Mayo Clinic Staff. “Sickle cell anemia.” MayoClinic.com. 2007. Web.
- Roberts, Michelle. “Network model predicts risk of death in sickle cell disease.” Boston University. 2007.