Volume of the Problem
Breast cancer is the most commonly diagnosed cancer and it is the leading cause of cancer mortality in women. Nearly 20% of breast cancer are familial, with 5 to 10% of cases being because of inherited gene mutation (hereditary). Approximately 65% of hereditary cases relate to BRCA1 or BRCA2 mutations (Breast cancer genes). The remaining cases of familial tendency are because of familial environmental factors or unrecognized gene mutations. Although more than 2600 mutations are identified on BRCA1 and BRCA2 genes, yet, the general characteristics of these mutations are; first, they are autosomal dominant. Second, their carrier frequency is nearly 1 per 800 individuals (in a white race) and may increase to 1 in 40 or 50 individuals in Ashkenazi Jews, and finally is that both genes are tumour suppressors. These gene mutations deregulate cell death and result in uncontrolled cell growth (Carroll et al 2008).
Origin of Breast Cancer and Genetic Link
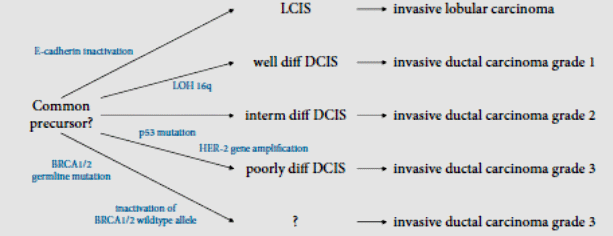
The majority of breast cancers arise from the epithelia that line the milk-producing lobules and ducts of the mammary gland. All females have a similar number of these cells, regardless of overall breast size. Therefore, breast size is an insignificant cancer risk factor. Approximately 80% of breast cancers are ductal in origin, between 5–10% are infiltrating lobular carcinomas and the remainder arise from diverse cell types. Like several common types of cancer, breast cancer begins with defined precursor lesions. Small, noninvasive lesions, typified by ductal carcinomas in situ (DCIS), are believed to progress to first invasive and then metastatic lesions. In contrast, small lesions found in the lobular epithelial, known as lobular carcinomas in situ (LCIS), are not believed to be precursor lesions that progress, although their appearance is associated with the subsequent disease. There are many risk factors for the development of breast cancer, the single most significant of which is a positive family history. More than 5% of breast cancers are thought to be hereditary in nature, arising as a consequence of germline alleles that confer cancer predisposition. Two high-penetrance tumour suppressor genes have been described, BRCA1 and BRCA2 (Sinn 2009). Figure 1 shows the multistep development of cancer breast and the pathway influenced by BRCA1 and BRCA 2 (Meijnen et al 2006, p. 113).
Several genes that are widely found to be mutated in other common cancers are also mutated in sporadic breast cancers. Nearly 30% of breast cancers harbor P53 mutations. More than 20% of breast cancers harbor somatically acquired mutations in PIK3CA and approximately 9% exhibit PIK3CA amplification. Other genes frequently amplified in breast cancers include CMYC and ERBB2 (also known as HER2/neu). Analysis of global gene expression in breast cancers has revealed numerous genes that are frequently over-expressed. C-SRC, CCND1 (a gene that encodes cyclin D), and BCL2 are examples of genes that have been reported to be expressed at high levels in many breast cancers. It is important to note that in most cases, the genetic alteration underlying the expression defect has not been determined. It, therefore, remains a possibility that overexpression of some genes in breast cancers is an indirect effect caused by deregulated upstream pathways, rather than a mutation at the locus in question (Woolcott et al, 2009).
Summary of Genetic Changes in Breast Cancer
Although relatively little is known about the molecular mechanisms leading to breast cancer development, breast cancers have probably been studied more than any other tumour type with regard to oncogene expression. MYC, ERBB2, or one of the RAS family has been found to be expressed in over 60% of cases. Mutations in highly penetrating susceptibility genes are estimated to underlie only 5–10% of breast cancers. However, these are important as individuals carrying such mutations are at very high risk of developing breast cancers, of having more than one primary cancer, and of passing these altered genes to their offspring. Following the genomic localization and subsequent identification of BRCA1 and BRCA2 (considered to be tumour suppressor genes) as breast cancer predisposition, the basic pattern of cancer risk associated with these genes has been defined (Bunz 2008).
Chromosomal Abnormalities and Potential Tumor Suppressors in Cancer Breast
In addition to the genetic alterations, there are a number of other reported alterations associated with breast cancer. LOH (loss of heterozygosity) on the long arm of chromosome 6 has been detected in 48% of primary breast cancers, confirming an earlier study indicating that this is a frequent occurrence in primary breast carcinomas. LOH for 7q31 may also be an early event in breast cancer development. Chromosome 1 abnormalities have also been reported to be a frequent occurrence and rearrangements of 1p correlate with a poor prognosis. Using DNA fingerprinting followed by LOH analysis it showed that regions at 2q21–24 and 6p21–23 may harbor novel tumor suppressor genes in breast cancer. It has also been shown that LOH at 2q21–24 is more frequent in high-grade than in low-grade tumours.
Chromosome 3p contains several tumour suppressor genes and 21 polymorphic and 2 non-polymorphic 3p markers were recently investigated by allele-typing and real-time PCR in a variety of epithelial malignancies, including breast cancer. A high frequency of LOH was found in many types of cancers, including breast cancer cases. It has been shown that the E-cadherin gene, responsible for the cell-cell adhesion was not mutated in some cases of infiltrative ductal or medullary breast carcinomas but was mutated in some other infiltrative lobular breast cancers using a PCR method. Although sample sizes of earlier studies were small; yet, they highlight the molecular explanation of how malignant cells spread in infiltrative lobular breast carcinoma. Evidence supports the involvement of Sigma 14–3–3 (a member of a class of molecular chaperones known to interact with cyclin-dependent kinases in breast cancer cells) in the neoplastic transformation of breast epithelial cells by virtue of its role as a tumour suppressor. Finally, there is evidence supporting retinoic acid receptor beta 2 (RAR 2) as a tumour suppressor. Bi-allelic inactivation of RAR 2 by epigenetic changes has recently been reported in breast cancer (Macdonald et al 2004).
Breast Cancer Tumor Suppressant Genes BRCA1 and BRCA2
There are two types of gene mutation that contribute to disease; first, germ-line mutations, which represent an accumulation of genetic changes in germ-line cells (cells carrying genetic material from one generation to another like ova). Second somatic mutations are generally acquired mutations that represent a change in DNA arrangement occurring after conception either spontaneously or because of exposure to mutants (Shanks et al 2008). BRCA1 and BRCA2 (breast cancer predisposition genes) are inherited germ-line gene mutations that encode multifunctional proteins. The mutant phenotype predisposes not only to breast cancer but also to ovarian cancer. BRCA1 and BRCA2 are large genes that span approximately 80 kb of genomic DNA. Both have a large central sequence of a gene’s DNA that transcribes into protein structures (exons) encoding >50% of the protein and both encode large proteins of 1863 (BRCA1) and 3418 (BRCA2) amino acids.
The molecular mechanism by which BRCA1 loss promotes tumor formation is unclear but it has been recently established that the BRCA1 RING finger region heterodimerizes with BARD1 and functions as an enzymatic mediator of protein ubiquitination. In normal cells, BRCA1 and BRCA2 are nuclear proteins and BRCA1 and BRCA2 mRNA and proteins are preferentially expressed during the late G1 (increase in cell size and preparation for DNA synthesis) and early S phase (DNA synthesis) of the cell cycle. BRCA1 and BRCA2 are expressed ubiquitously and in mouse embryos, they are highly expressed during mammary epithelial proliferation and differentiation. Expression of these messages, in the mammary gland, is developmentally controlled. Further, it decreases during lactation but is provoked during puberty and pregnancy. Null mutations in both genes result in embryonic lethality. Figures 2 and 3 show the domain structure of BRCA1 and BRCA2 (Boulton 2006).
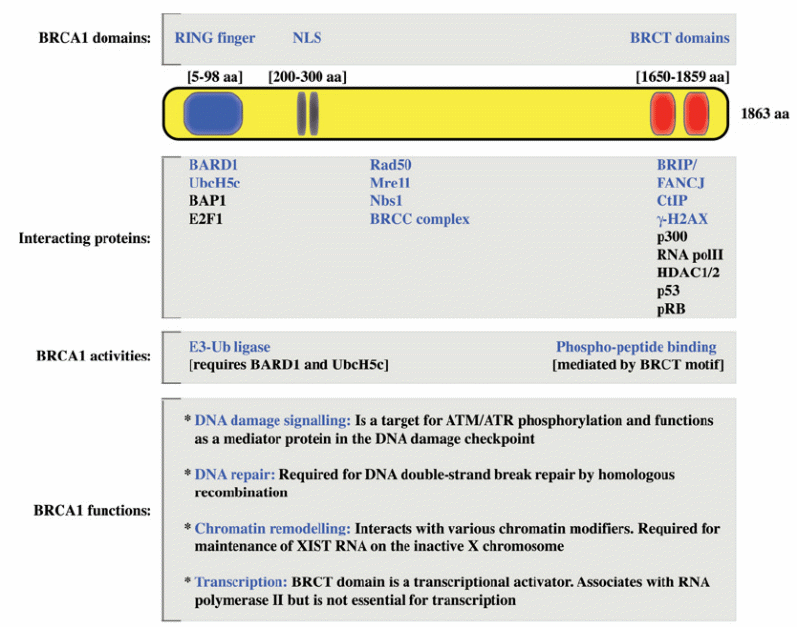
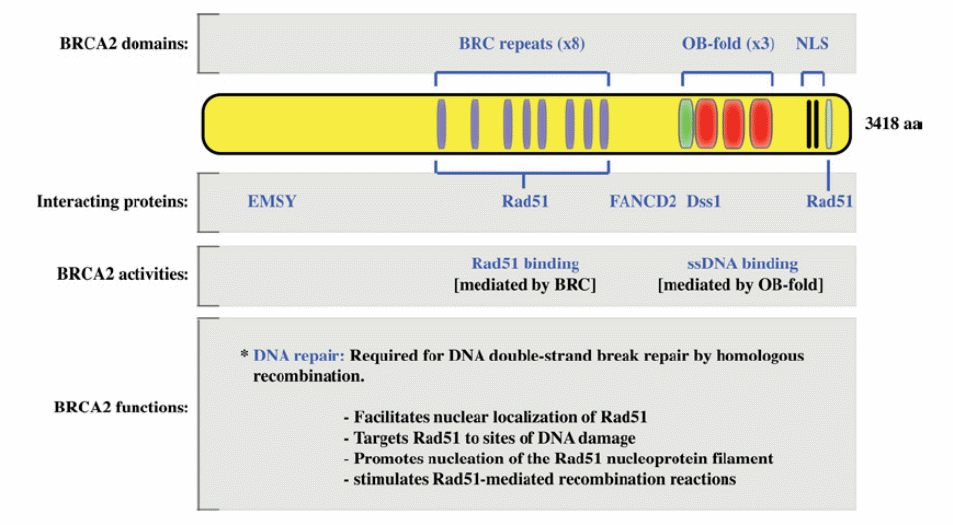
The biochemical interaction of BRCA1 and BRCA2 with proteins involved in DNA repair suggests their role in homologous recombination (HR). A striking connection between double-strand break repair defects and oncogenesis has been shown for BRCA1 and BRCA2. Both proteins interact with RAD51 protein which catalyzes structural change as an early step in homologous recombination that results in the formation of heteroduplex molecules. BRCA1 and BRCA2 are essential for normal development as patients in whom both alleles of BRCA1 or BRCA2 are mutated have not been found. Evidence suggests the role of BRCA1 in DNA repair is more expansive than that of BRCA2 and involves many pathways. BRCA1 operates in the signalling of DNA injury and repair through homologous recombination (exchange of nucleotide sequences between two comparable or matching strands of DNA). It also helps nucleotide excision repair through repairing DNA double-strand breaks (non-homologous). BRCA2 adjusts the activity of RAD51 needed for homologous recombination. Recent research evidence ascertained that BRCA2 operates directly in homologous recombination, which provides insight on the loss of recombination-mediated double-strand break (DSB) repair observed in BRCA2-associated cancers. These findings have prompted searches for alterations of RAD51 in patients with breast tumours. In one such study with patients with early-onset breast cancer, no germ-line mutations were identified although in another study a single nucleotide polymorphism (SNP) was found in the 5′ UTR region of RAD51 which was associated with an increased breast cancer risk in BRCA2 carriers (Lin et al 2009).
Both BRCA1 and BRCA2 appear to be involved in DNA repair pathway networks. It has been found that BRCA1 contains a peptide domain called BRCT that appears to be a common motif in other proteins involved in DNA repair. BRCT domains bind phosphopeptides in protein-binding partners and typically occur as 80–100 amino acid sequences present as tandem repeats in BRCA1. These sequences recognize substrates phosphorylated by the DNA repair kinases ATM and ATR in response to γ-irradiation. A mutation in the BRCT domain of BRCA1, which prevents binding to phosphopeptides, may explain why this mutation predisposes women to breast and ovarian cancer. Research showed that the BRCA1 BRCT domain binds a phosphorylated, BRCA-associated, DNA repair helicase. This interface is cell cycle-regulated and needed for DNA damage-induced checkpoint control (related to biochemical conduits ending the delay or arresting cell-cycle progression) of the G2 to M cell cycle phase transition. Therefore, it is suggested that BRCT ambit-containing proteins are involved in DNA repair and checkpoint control of the cell cycle (Ruddon 2007, p. 366).
An association between a heterozygous BRCA1 or BRCA2 mutation in human cells and radiosensitivity has been demonstrated recently. Although data may not provide sufficient evidence to prove a causal relationship between heterozygous BRCA mutations and radiosensitivity they are important as this phenotype could possibly predispose to an increased rate of radiation-induced mutagenesis and carcinogenesis (Capalbo et al 2006).
Somatic mutation of p53 is frequent in human BRCA1 and BRCA2 breast tumours, and many BRCA-associated p53 mutations are novel. Genetic instability caused by the loss of BRCA1 or BRCA2 could trigger mutations, including mutations in checkpoint genes such as p53. Thus, mutant p53 seems to trounce incomplete proliferation and pilot uncontrolled proliferation resulting in invasive growth. Brisk multiplication of breast epithelial cells at times of pregnancy and menarche provides a ground for a somatic mutation that may involve loss of a BRCA1 or a BRCA2 allele. Tissue specificity of BRCA1 and BRCA2 tumourigenesis might be attributable to both the hormonal environment of breast (and ovarian) cells, as well as to the tissue-specific expression of as-yet-unknown genes. BRCA1- and BRCA2-associated human breast cancers have different morphological and immunohistochemical characteristics, including different gene expression profiles. Importantly, from a therapeutic viewpoint, restoring BRCA1 or BRCA2 function would not be useful after checkpoint mutations had occurred (Mann et al 2006).
An important recent finding has been that an apparent BRCA1 homozygous knockout was found to be an artefact of the PCR. This suggests that mispriming due to inequitable primers at the site of SNPs results in discriminatory elaboration of one allele. This may represent a considerable fraction of cases of mutation detection insensitivity. These results have important cautionary implications for all PCR-based mutation detection methods used in research and in particular in a clinical genetic laboratory (Schwartz et al 2009).
Conclusion
To date, BRCA1 and BRCA2 are the two major genes identified that link to breast cancer high risk. Based on the above discussion, a logical inference is BRCA-BARD1 has a role in DNA repair and checkpoint signaling. Evidence suggests BRCA2 has a role in homologous recombination, although how BRCA2 function is adjusted in response to DNA damage is yet to clarify. Using rapid and reliable testing to identify females with specific BRCA mutations can improve management strategies.
Reference List
Boulton, S. (2006). Cellular functions of the BRCA tumor-suppressor proteins. Biochemical Society Transactions, 34(5), 633-645.
Bunz, F. (2008). Chapter 6, Genetic Alterations in Common Cancers. In Bunz, F. (Ed.), Principles of Cancer Genetics. (pp. 227-258). New York: Springer.
Capalbo, C., Ricevuto, E. & Vestri, A. et al. (2006). BRCA1 and BRCA2 genetic testing in Italian breast and/or ovarian cancer families: mutation spectrum and prevalence and analysis of mutation prediction models. Annals of Oncology, 17(Suppl. 7), vii34-vii40.
Carroll, J. C., Carol, C. & Judith, A. et al. (2008). Hereditary breast and ovarian cancers. Canadian Family Physician, 54, 1691-1692.
Lin, Y. P., Chen, Y. L. & Chang, H. T. et al. (2009). Nature of genetic variants in the BRCA1 and BRCA2 genes from breast cancer familiea in Taiwan. Life Science Journal, 6(3), 99-104.
Macdonald, F., Ford, C. H. J. & Casson, A. G. (2004). Molecular Biology of Cancer. 2nd ed. Bios Scientific Publishers: London.
Mann, G. J., Thorne, H. & Balleine, R. L. et al. (2006). Analysis of cancer risk and BRCA1 and BRCA2 mutation prevalence in the kConFab familial breast cancer resource. Breast Cancer Research, 8(1), R12-R18.
Meijnen, P., Peterse, J. L. & Vijver, M. J. V. D. (2006). DCIS: Pathology and Molecular Markers. In Piccart, M. J., Wood, W. C. & Mien, H. C. et al (Eds.), Breast Cancer and Molecular Medicine. (pp. 99-124). Berlin: Springer.
Ruddon, R. W. (2007). Cancer Biology. 4th ed. New York: Oxford University Press.
Schwartz, G. F., Hughe, K. S. & Lynch, H. T. et al. (2009). Proceedings of the International Consensus Conference on Breast Cancer Risk, Genetics, & Risk Management, April, 2007. The Breast Journal, 15(1), 4-16.
Shanks, M. E., May, C. A. & Dubrova, Y. E. et al. (2008). Complex germline and somatic mutation processes at haploid human minisatellite shown by single-molecule analysis. Mutation Research, 648, 46-53.
Sinn, H. P. (2009). Breast cancer precursors: lessons from molecular genetics. J Mol Med, 87, 113-115.
Woolcott, C. G., Maskarine, G. & Haiman, C. A. (2009). Association between breast cancer susceptibility loci and mammographic density: the Multiethnic Cohort. Breast Cancer Res, 11(1), R10-R11.