Summary
The objective of the experiments was to remove the EGFP gene from the pEFGP vector and to clone in another vector, pET28b, derived from bacteriophage T7 vectors. The cloned gene would be under T7 RNA polymerase promoter control. Thus, the fluorescent gene would be expressed in larger quantities and would enable us to construct an improvised expression vector for applications. The advantage with both pEFGP and pET28b vectors is that both have one digestion site each for restriction enzymes, Eco R1 and Bam H1. Upon digestion of the vectors, while pET28b would just open into a linear plasmid, the EGFP gene would be released from pEFGP.
Such DNA fragments can be detected by agarose gel electrophoresis and subsequently eluted, cleaned, and then EGFP can be ligated with linearized pET28b to give pET28bEGFP expression plasmid construct. This would be transformed into two competent cells of E. coli and subsequently selected for the transformed cells. The experiments are aimed to successfully construct pET28bEGFP plasmid and to screen its presence in transformed cells.
Introduction
Several cloning vectors use fluorescent marker proteins derived from bio-fluorescent jellyfish, Aequorea victoria. Among them are the green fluorescent protein (GFP) and its yellow-green variant, YFP, whose cDNA has been cloned into pUC derivative, pPD16.43, to yield a recombinant vector pEGFP (enhanced GFP). Such vectors carry the ampicillin resistance marker for sorting out the Escherichia coli cells harboring the plasmid on ampicillin-containing plates.
Once these vectors are expressed in transgenic eukaryotic cells, like animal cell lines, the transformed cells would emit at 510 nm bright green light under flow cytometry if they are excited with 488 nm light. Any foreign gene(s) can be co-transformed with pEGFP, and the expression of that gene can be monitored. One such application is to elucidate Hepatitis B virus-induced alteration in expression of genes leading to carcinoma in isolate human bile duct cells.
Results
The vectors (pET28b) (V) and (pEGFP) (I) were separately digested with Bam H1 in the presence of restriction digestion buffer and ddH2O. The reaction mixture composition is given in Table 1. The DNA was allowed to be digested for 30 min at 37oC and brought to room temperature. The Bam H1 digested V was designated as VB and I as IB DNA. Aliquots of these DNAs were then digested in the same reaction mixture with Eco R1 for an extended period of 1 h.
The resulting double digested DNAs were respectively labeled VBE and IBE. The success of restriction digestion was examined on 1% agarose gel containing SYBR green dye, using a submarine gel electrophoresis apparatus. This harmless dye intercalates with DNA and fluoresces green/orange under UV. The gel and sample buffer were prepared using Tris-acetate-EDTA buffer. The DNA samples (VB, VBE, IB, IBE, the uncut plasmids uV and uI, and DNA ladders, Lambda BstEII and 1 KB plus standards) were mixed with tracking dye and applied to the gels. Upon staining, the bands of DNA were visualized and photographed.
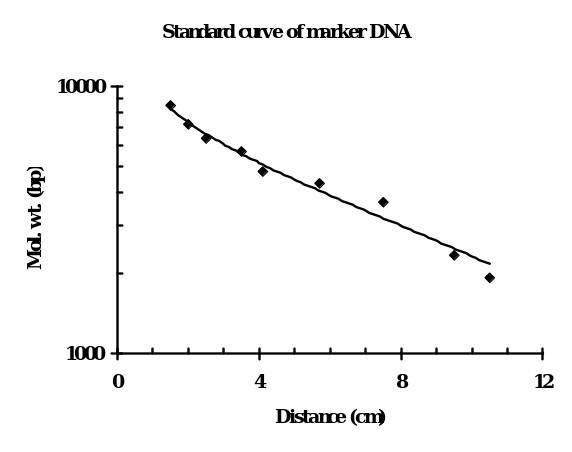
The bench 4 gel picture, as shown in Fig. 1 (right), reveals the presence of (from left to right) 1 band (ul), 1 band (IB), 2 bands (IBE), and 1 band each of uV, VB and VBE, along with the Lambda BstEII ladder (ca. 9 bands) and 1 KB ladder (ca. 13 bands). Fig. 1 (left) illustrates our bench 6 gel picture in which only IB, IBE, VB, and VBE bands were visible, and neither the intact plasmids nor the standards were visible. For determining the molecular size (in bp) of the linear DNA, i.e., the fragments that were generated upon restriction digestion, the following consideration was made: distance traveled by the DNA is inversely proportionate to the log10 of molecular weight. For roughly estimating the molecular size in bp of the DNA, the distance traveled by each ladder (fragment) of the standard DNA from the leading edge of the start point was plotted against their know size (bp) using semi-log graph paper.
The minimum size of the fragment in the case of Lambda BstEII ladder is 702 bp, which is not visible in the gel. The bands were discernable from the third fragment that is 1929 bp onwards. The distance of the bands of unknown bands (in cm) was matched, and accordingly, the molecular weights were estimated. For positions in between the two adjacent ladders, an approximation of in-between weights was taken into consideration.
This calibration is only for the linear DNA and can not be equated with the circular DNAs like uI and uV, which are intact plasmids. The standard curve of Lambda BstEII standard DNA is shown in Fig. 2. Accordingly, the size of each fragment(s) in the lane was extrapolated. Based on such calculations, the apparent molecular weight(s) in bp of the fragments in lanes were as follows: IB = 6,300, IBE = 3,600 and 5,700, VB = 8,500 and VBE= 8,500. This indicates that only two bands of unequal size were retrieved upon double digestion of I (IBE).
One of the fragments in IBE (the smaller one) is apparently the gene of interest that is the EGFP gene. The IBE sample was re-electrophoresed as before on identical conditions, and again, bands of a small and a large fragment were resolved. Using a sharp spatula, which was first rinsed with water and then with ethanol, agarose around the small band’s position was excised. The agarose piece weighed approximately 180 mg.
To this, we have added 1.8 µL of GENECLEAN Turbo Salt (GTS) in Tube I. Likewise, the V (vector) sample was eluted from the gel and cleaned as above. For V processing, to 15.5 µL DNA sample, 500 µL GTS was added. After the clean-up operations using the GLEANCLEAN spin columns, we recovered 34 µL of pure V and 30 µL of pure I. For the cloning reaction, the pure I DNA (EGFP cDNA) has to be ligated using T4 ligase with open linearized V DNA (pET28b). The following additions were made:
- T4 DNA ligase, __1__ units/µl, 1 µl in a tube marked “T4”, and
- 5X Ligase buffer: ___4 µl___ in tube marked “5X”
The ligation mixture composition (B4 information) is given in Table 2. It is important to include a control set without ligase. pDRAW 32 DNA analysis software was used to “a” determine the size of the insert DNA that we expect to clone into pET28b, “b” to construct the new pET28b-EGFP and determine its size, and “c” to draw a picture showing all the possible products that we could get for the two ligation tubes we set up. Fig. 3 depicts all the possible products with their size of DNA. Here, uncut vector pET28b (5368 bp), vector with insert EGFP cDNA (6123 bp), and vector with the complete vector part of pEGFP, called pET28b, BackbonepEGFT (7956 bp) are shown.
The entire pET28b.EGFP or pET28b and EGFP after ligation was then transformed in two E. coli strains. Either these transformational bacteria were made competent by heat shock treatment, or the DNA uptake was facilitated by electroporation. The DH5α-E cells were obtained from a commercial source and were transformed by ligation mixture using the electroporation method. The other strain, BL21 (DE3), was transformed by the heat-shock method.
For pursuing the electroporation experiments, 200 µl of Elecromax DH5α-E cells were thawed in an ice bath. Two microcentrifuge tubes (Eppendorf) labeled “O” and “E”, electroporation cuvette, and culture tubes containing SOC medium (2%(w/v) back to tryptone, 0.5% (w/v) yeast extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, and 10 mM MgSO4; pH~ 6.7-7.0) were chilled. First, 100 µl thawed cells were transferred to the marked microcentrifuge tubes.
From the microcentrifuge tube marked “O,” an aliquot was transferred to the bottom of a chilled cuvette using a sterile Pasteur pipette. The ice/water was wiped out completely from the cuvette, which was then transferred to the cuvette holder of the electroporator. The electroporation was carried out at 2.5 kV. At a time constant of 5, 500 µl was added to the cuvette and immediately shifted to an ice bath. Subsequently, the cells were transferred to a culture tube marked “O” placed in ice containing 1 ml SOC medium. The above procedure of electroporation was also carried out by pipetting out cells from a microcentrifuge tube marked “E,” and to this, 1 µl of pET28bEGFP DNA was added.
The cells and DNA mix were transferred to a new electroporation cuvette, and electroporation was performed as above. In the end, the cells will be transferred to a culture tube marked “E” containing SOC medium. Placing the tubes at 37oC for 60-90 minutes would allow the expression of antibiotic (kanamycin) resistant genes. The recovered cells from each tube were then plated on an LB agar medium with kanamycin and iso-propylthiogalactoside (IPTG).
The natural transformation of heat-shocked BL21 (DE3) was carried out using a set of controls and samples, as shown in Table 3. The cells, to which DNA was given, were incubated first in ice for 30 min, and then heat shock treatment was given. These were diluted with 1 ml LB medium and were shaken for 45-60 min at 37oC before being plated on LB medium containing kanamycin (KAN) with or without IPTG and ampicillin (AMP).
Table 4 shows the plate set-up for bacterial plating. As can be seen, cells + linearized pET28b + EGFP DNA + ligase (Tube 1) and cells + linearized pET28b + EGFP DNA – ligase (Tube 2) sets were plated on KAN +/- IPTG and AMP plates, cells + pET28bEGFP (positive controls; Tube 3) were plated on KAN +/- IPTG plates, and cells without any DNA (negative controls; Tube 4) were plated on only KAN – IPTG plates. The plates were then wrapped and kept upside down at 37oC for a maximum of 18 hours. What we expect to see on the plates from both the transformation experiments (electroporation and heat shock transformation) is shown in Fig. 4.
No colony was seen in cells taken from Tube 2, in which cells were incubated without ligase. White colonies that should glow green light under black light can be seen in Tube 1 (cells with ligase), but only with KAN +/- IPTG and not AMP. We are supposed to see similar colonies in Tube 3 cells (positive controls) as well (except AMP as this set was not kept). Finally, for the cells in Tube 4, we should not see any cells other than contamination (if any). In Tube E (cells with plasmid DNA, electroporation) and not in Tube O (cells without DNA, electroporation), we should see colonies in the presence of KAN + IPTG. In the previous case, without IPTG, no colony should be detected.
Table 1: Restriction enzyme digest.
Table 2: Ligation mixtures.
Table 3: (5.1). Transformation of competent cells.
Table 4 (5.2): LB agar plate set-up for bacterial plating.



Discussion:
Addressing the five questions each in one paragraph.
- We expected that pEGFP and pET28b would digest with Eco R1 and Bam H1. This is because both the vectors have one site each for the two restriction enzymes. Therefore, the intact, uncut vectors (molecular weights = 5,368 bp for pET28b, and 2,588 bp for pEGFP, calculated from pET28b.BackbonepEGFP – pET28b; Fig 3) would resolve in the gel as single bands, and upon digestion, with Bam H1 they would linearize and again give single bands. Once the linearized DNA’s were re-digested with EcoR1, we expected two bands in the case of pEGFP as it has a distinct site for Eco R1 away from Bam H1 cut site. Thus the double digested plasmid should have a small fragment of the EGFP gene, sandwiched between the two enzymes’ sites, and a large fragment of the rest of the plasmid. On the other hand, double digested pET28b is expected to give the identical length of the fragments, whether cut by Bam H1 alone or along with Eco R1. This is because the two sites are very close and are rather difficult to differentiate by electrophoresis. Calibration curves prepared for standard DNA markers like Lambda BstEII or 1 KB, by plotting the fragment size and the distance traveled by each fragment should be linear. The smaller of the two bands in the IBE slot is expected to be of the EGFP gene, which would be ligated with open pET28b. As we were cloning the EGFP gene directly in pET28b, which has a kanamycin resistance gene under the control of IPTG-inducible β-gal promoter, only the transformed cells (positive controls), with pET28b.EGFP construct would grow on KAN + IPTG plates and glow green under the dark field. Obviously, the cells which received no DNA would not express both the genes. Only the pEGFP vector has a resistance marker for selection in AMP plates, and as we were not working with this vector, we would not have expected any colony on AMP plates.
- In accordance with the expectations, the electrophoresis of pET28b (V) and pEGFP (I) single digest with Bam HI gave one band each (IB, VB), indicating one restriction site for this enzyme. Indeed, we found one band each for the uncut and the Bam H1 cut vectors. The unexpected finding was that upon Bam H1 digestion, the resulting molecular weights of the linearized plasmids were higher than the putative weights of intact plasmids (IB = 6,300 bp and VB = 8,500 bp, respectively). As can be seen in Fig. 1 bench 4 (IBE slot), we observed two distinct bands representing two cuts, with molecular weights of 3,600 bp and 5,700 bp. This smaller band, being the EGFP gene, was later confirmed by cloning and expression of this gene in the pET28b vector. Upon double digestion of pET28b, the fragment of size 8,500 bp was identical to the fragment obtained from a single digest (Fig. 1, bench 4). Once the log10 of the molecular weights of the Lambda ladder was plotted against the distances traveled by corresponding fragments from the well, we found points which, upon regression, gave a straight line (Fig. 2). In the transformation experiments, all incubations in which DNA was omitted failed to produce cells growing on KAN +/- IPTG, suggesting the cells do not have inherent kanamycin resistance. Only the positive controls exhibited expression of KAN and EGFP (Fig. 4). Ligase omitted sets also failed to grow on KAN plates. This suggests that prior sealing of DNA construct is required before being transformed. No growth on AMP plates confirmed that there was no contamination of pEFGP. This possibility would have existed. Had it been a mistake to collect the larger band in the IBE slot, which was of the remaining pEGFP after removal of the EGFP gene? This DNA has the marker for ampicillin resistance.
- The difference in molecular sizes of uncut and Bam H1 cut vector DNA’s (Fig 1) was from the fact that supercoiled nature of closed plasmids enables them to migrate farther compared to linear DNA of identical size. Due to invisibility, some of the bands of the 1 KB ladder could not be properly deduced. The calibration curve for the 1 KB standard also could not be drawn as the lower bands (100-1000 bands) were missing, and the band positions were not coinciding with the available information. In transformation experiments, the results with “E” cells were rather strange. While colonies did appear in KAN + IPTG plates as expected from such cells, the cells did not fluoresce green under dark background. The most likely explanation is that in DH5α-E cells, the turnover of either the mRNA of EGFP or protein itself is higher than in BL21 cells. The rapid degradation of the transcript or the protein after expression may result in failure in the detection of green fluorescence in such cells, even though they may grow in KAN plates.
- The Lambda BstEII ladder resolved but with lower bands missing. Either the DNA quantity was too low, or the bands were lost from the bottom of the gel. It would be a better option to apply larger volumes of this marker until we see the lower bands. Alternatively, agarose concentration can be slightly increasing (1.1-1.2%). Further, it is advised to check the authenticity of the 1 KB ladder and re-apply higher volumes so that all fragments are discernable to draw a curve. The unusual results of cells “E” can be verified. For this, we have to carry out Northern blot analysis of EFGP transcript and Western blot analysis of the protein. The relative amounts of the two species in IPTG induced DH5α-E and BL21 strains as against non-induced cells would generate data to suggest whether they are degraded fast in a particular E. coli strain than the others.
- We recommend the replacement of ethidium bromide by SYBR green. This experiment gave caution towards the use of strategy and strains of E. coli to be used for expression of animal reporter genes, like EFGP. This also indicates that the natural method of transformation is superior as compared to electroporation in the expression of recombinant animal proteins, such as the FGP. We shall recommend using this method as it is also cheap and can be done conveniently in any lab.
Answers to Questions
Why do we incubate the vector, pET28b, and the insert, pEGFP, with both Eco RI and Bam HI?
Both vectors have one site each for Eco R1 and Bam H1, and by double digestion, the plasmids were linearized with corresponding overhangs, which would not re-ligate. Thus we would always get insert being ligated. This was the objective.
Why did we incubate with Eco RI and Bam HI in intervals?
Eco R1 and Bam H1 treatments were done at intervals because the treatment conditions were different. Bam H1 treatment was for 30 min and Eco R1 for 1 hour, not possible to give simultaneously. Any difference in time would either incompletely digest or over-digest, thus producing artifacts in electrophoresis.
Why did we remove some of the samples after treatment with Bam HI?
This was done for IB, and VB samples which were only Bam H1 digested vectors and were electrophoresed separately from IBE and VBE.
What important information can we gain from looking at the samples cut with Bam HI alone?
We get two pieces of information,
- the two vectors were linearized as Bam H1 has a single site in both the plasmids and
- the apparent molecular weight of the linearized DNAs is more than the supercoiled circular plasmids.
Is it possible to predict how often a restriction enzyme will cut a particular piece of DNA? Why or why not?
Yes, it is possible by checking the change in migration of closed and opened DNAs. The digestion can be repeated at different time intervals, and once we get the clear difference in migration and no additional artifacts, we consider the treatment as being standard and predictable.
Did Bam HI cut both pEGFP and pET28b to completion?
Yes, Bam H1 cut both the pEGFP and pET28b vectors completely. This conclusion is based on the fact that the linearized DNA thus obtained (IB, VB) migrated to a lesser distance than the closed plasmids (I, V).
Explain why we used two DNA standards to assess the quality of the plasmid digest.
Apart from BstEII cut Lambda DNA with already known standard fragments, we have used 1 KB plus a standard marker. This ladder is supposed to give bands in the range of 12,000 to 100 bp. Two ladders are normally used to get a narrower range of standard molecular weights to deduce more precisely the size of the fragments. If, in this case, the fragment size were <700 bp or >10,000, BstEII cut Lambda would have been useless, and for this, 1 KB plus would be more appropriate.
Why can you not use uncut DNA references to estimate the size of your DNA fragments?
Uncut DNA references (circular) would be supercoiled and migrate farther to the corresponding cut (linear) DNA. If it is nicked, then it would migrate less than the same size linear DNA. Hence both can not be equated. We ran the uncut plasmid DNA to ensure that the fragments after restriction digestion came from these sources.
In the lab, we have increased the efficiency of our ligation of the pET28b vector with EGFP insert by purifying the DNA fragment containing the EGFP cDNA from the original pUC vector (pPD16.43) that it was in. Why do we do this? Why would doing this increase the probability of getting the product we want?
Yes, the EGFP insert in the original pUC vector (pPD16.43) is expressed under low expressible Plac (Lac promoter), and once it is cloned in pET28b, the insert is expressed with over-expressible T7 RNA polymerase promoter, and hence the copy number of transcript increases and thus the protein. We should expect more product formation.
We had a control ligation tube “without ligase.” What other controls might be important to do for the ligation reaction and why?
One negative control in which DNA (one of both fragments to be ligated, or both) is omitted would be required so as to minimize any possibility of self-ligation. This problem will not arise in the present experiment as we conducted directional cloning.
What is “directional cloning,” and how did we achieve this using both BamHI and EcoRI digestions of our original plasmid DNAs?
Directional cloning means where we keep a special orientation of the insert. Normally the coding region of the gene (5′ – 3′), which is to be expressed, is placed after promoters in the vector so that the correct transcript is formed and protein expression is normal. In this experiment, by using Bam H1 and Eco R1 double digestions, we have produced two different overhangs, one each for the enzyme. Likewise, the insert also had double digestion, thus yielding Bam H1 and Eco R1 overhangs. As only the correct overhangs will pair, e.g., Bam H1 with Bam H1 and Eco R1 with Eco R1, there would not be a mismatch, and the insert will be placed in only one direction and not in the opposite. Ligation will seal the gaps, and the correct kind of protein will be expressed.
Ligase will not work on a 5’OH adjacent to a 3’OH. You can produce 5′ OH ends by treating linear DNA with Calf Intestine Alkaline Phosphatase (CIAP). This enzyme removes phosphate groups from DNA. What would the ends of the DNA molecules look like if a CIAP-treated vector that has been cut with Eco RI were ligated with an insert DNA molecule that has Eco RI sticky ends?
The situation is that Eco R1 treated vector is further treated with CIAP to remove 5′ phosphate to give 5’OH. Naturally, the 3’OH is intact. The insert has normal 3′ and 5′. As sealing has to take place at four positions, two on either side of the insert so long as normal 3′ and 5′ ends are available, in this case, only the intact 3′ of the vector will ligate with intact 5′ of the insert, and this will happen only at one of the two stands at both the ends. This will result in one nick each at two ends of the insert.
Use the pDRAW 32 DNA analysis Software on the lab computers or NEBcutter to:
- Determine the size of the insert DNA that you expect to clone into pET28b.
- Construct the new pET28b-EGFP and determine its size.
- Draw a picture showing all the possible products that we could get for the two ligation tubes you set up.
The sizes are as follows:
- Insert DNA: 6123 – 5368 = 755 bp;
- pET28b.EGFP = 6123 bp;
- Diagrams already given under Results section (Fig. 3).
Why did we plate the cells transformed with the ligation mixtures on AMP plates? Do you expect to see colonies on the AMP plates? Why?
We use AMP plates though we do not expect any growth. pEGFP vector has an ampicillin resistance marker, and we used this plasmid to get the EGFP gene. As we have worked with pEGFT and also have electrophoresed this plasmid and a fragment generated after removal of EGFP, there is a slim possibility that this marker may be accidentally retrieved in the excision and clean-up steps. Such ligated DNA in E. coli may express AMP, and thus we have a control to exclude this possibility.
Will all colonies that arise on the Kanamycin plates from tube #1 contain the plasmid we are trying to make? What are other possibilities?
Yes, because pET28b will not re-ligate as the two overhangs are from different restriction enzymes. The only plasmid of our interest will transform in this set.
What is the purpose of using DNA from tube #2, the ligation reaction that did not contain ligase to transform cells? In a perfect world, what do you predict that you would see from plating these cells transformed by DNA from tube #2? What kind of colonies may grow on the KAN plates with transformed bacteria from tube #2?
There is a small possibility that the sticky ends pair, and without ligation, the plasmid enters the cells. Upon entry by the action of E. coli ligase and/or nick translation by DNA Pol I, the gaps are sealed. In this case, we shall see the KAN marker in the transformed cells.
Would we expect DH5 α -E transformed with the positive control to express EGFP when grown on KAN plates with IPTG? Why or why not?
It may or may not, depending on the turnover of the transcript or the protein. In this experiment, expression of EGFP was not found even though they were selected on KAN. Probably the GFP was degraded in such transformed cells.
References
Lewin, Benjamin. Genes VII. New York: Oxford University Press, 2000.
Sambrook, Joseph, and Davis W. Russel. Molecular Cloning – A laboratory manual (3rd Ed.). New York: Cold Spring Harbor Laboratory Press, 2001.
Zou, Sheng-Quan, Zhen-Liang Qu, Zhan-Fei Li, and Xin Wang. “Hepatitis B virus X gene induces human telomerase reverse transcriptase mRNA expression in cultured normal human cholangiocytes.” World Journal of Gastroenterology 10(15) (2004): 2259-2262.